

Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1
- PMID: 19901262
- PMCID: PMC2808162
- DOI: 10.1182/blood-2008-12-196840
Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1
Abstract
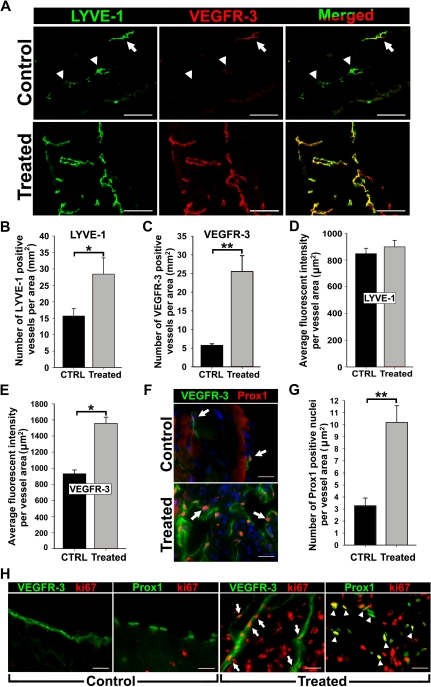
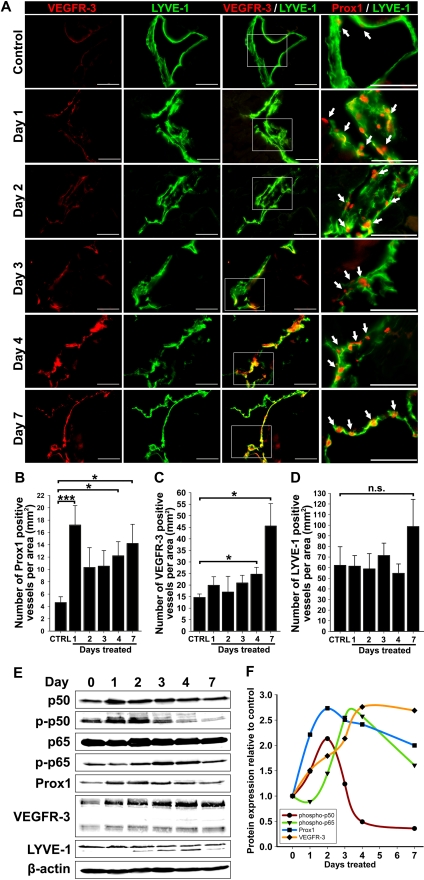
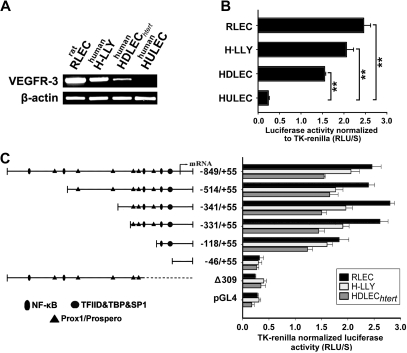
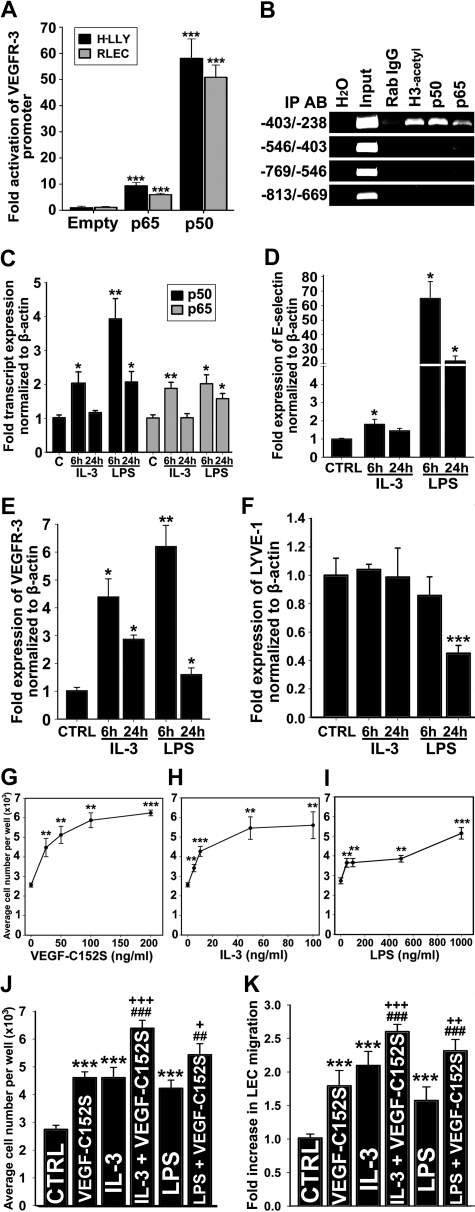
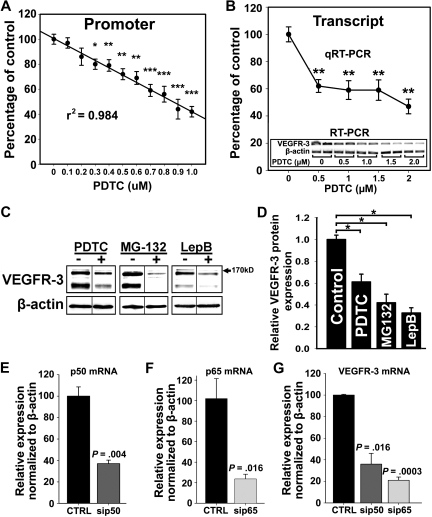
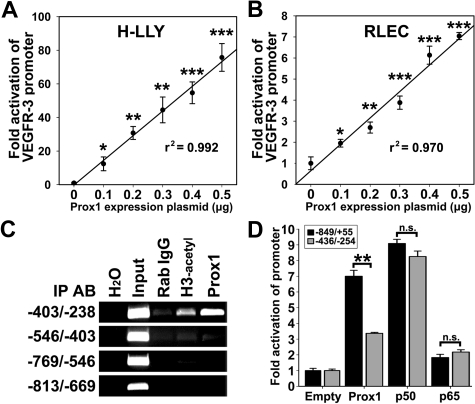
The concept of inflammation-induced lymphangiogenesis (ie, formation of new lymphatic vessels) has long been recognized, but the molecular mechanisms remained largely unknown. The 2 primary mediators of lymphangiogenesis are vascular endothelial growth factor receptor-3 (VEGFR-3) and Prox1. The key factors that regulate inflammation-induced transcription are members of the nuclear factor-kappaB (NF-kappaB) family; however, the role of NF-kappaB in regulation of lymphatic-specific genes has not been defined. Here, we identified VEGFR-3 and Prox1 as downstream targets of the NF-kappaB pathway. In vivo time-course analysis of inflammation-induced lymphangiogenesis showed activation of NF-kappaB followed by sequential up-regulation of Prox1 and VEGFR-3 that preceded lymphangiogenesis by 4 and 2 days, respectively. Activation of NF-kappaB by inflammatory stimuli also elevated Prox1 and VEGFR-3 expression in cultured lymphatic endothelial cells, resulting in increased proliferation and migration. We also show that Prox1 synergizes with the p50 of NF-kappaB to control VEGFR-3 expression. Collectively, our findings suggest that induction of the NF-kappaB pathway by inflammatory stimuli activates Prox1, and both NF-kappaB and Prox1 activate the VEGFR-3 promoter leading to increased receptor expression in lymphatic endothelial cells. This, in turn, enhances the responsiveness of preexisting lymphatic endothelium to VEGFR-3 binding factors, VEGF-C and VEGF-D, ultimately resulting in robust lymphangiogenesis.
Figures

References
-
- Swartz MA, Hubbell JA, Reddy ST. Lymphatic drainage function and its immunological implications: from dendritic cell homing to vaccine design. Semin Immunol. 2008;20(2):147–156. - PubMed
-
- Shin WS, Rockson SG. Animal models for the molecular and mechanistic study of lymphatic biology and disease. Ann N Y Acad Sci. 2008;1131:50–74. - PubMed
-
- Jamieson T, Cook DN, Nibbs RJ, et al. The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol. 2005;6(4):403–411. - PubMed
-
- Achen MG, Stacker SA. Molecular control of lymphatic metastasis. Ann N Y Acad Sci. 2008;1131:225–234. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
