

RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction
- PMID: 19901339
- PMCID: PMC2785285
- DOI: 10.1073/pnas.0905686106
RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction
Abstract
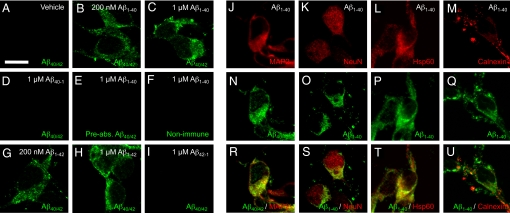
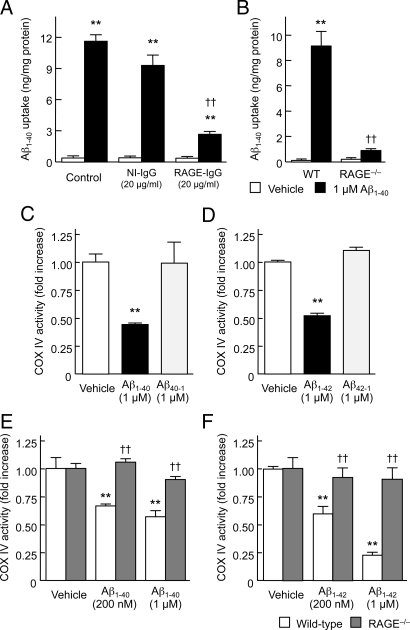
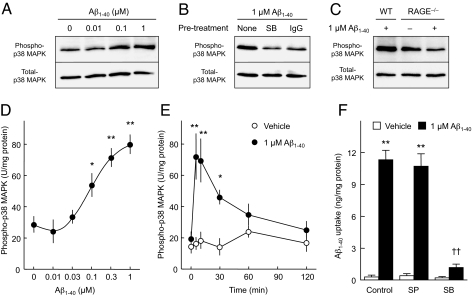
Intracellular amyloid-beta peptide (Abeta) has been implicated in neuronal death associated with Alzheimer's disease. Although Abeta is predominantly secreted into the extracellular space, mechanisms of Abeta transport at the level of the neuronal cell membrane remain to be fully elucidated. We demonstrate that receptor for advanced glycation end products (RAGE) contributes to transport of Abeta from the cell surface to the intracellular space. Mouse cortical neurons exposed to extracellular human Abeta subsequently showed detectable peptide intracellularly in the cytosol and mitochondria by confocal microscope and immunogold electron microscopy. Pretreatment of cultured neurons from wild-type mice with neutralizing antibody to RAGE, and neurons from RAGE knockout mice displayed decreased uptake of Abeta and protection from Abeta-mediated mitochondrial dysfunction. Abeta activated p38 MAPK, but not SAPK/JNK, and then stimulated intracellular uptake of Abeta-RAGE complex. Similar intraneuronal co-localization of Abeta and RAGE was observed in the hippocampus of transgenic mice overexpressing mutant amyloid precursor protein. These findings indicate that RAGE contributes to mechanisms involved in the translocation of Abeta from the extracellular to the intracellular space, thereby enhancing Abeta cytotoxicity.
Conflict of interest statement
Conflict of interest statement: D. Stern is a consultant for TransTech Pharma.
Figures

References
-
- Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. - PubMed
-
- LaFerla FM, Oddo S. Alzheimer's disease: Aβ, tau, and synaptic dysfunction. Trends Mol Med. 2005;11:170–176. - PubMed
-
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
