

Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury
- PMID: 19902481
- PMCID: PMC3164814
- DOI: 10.1002/hep.23365
Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury
Abstract
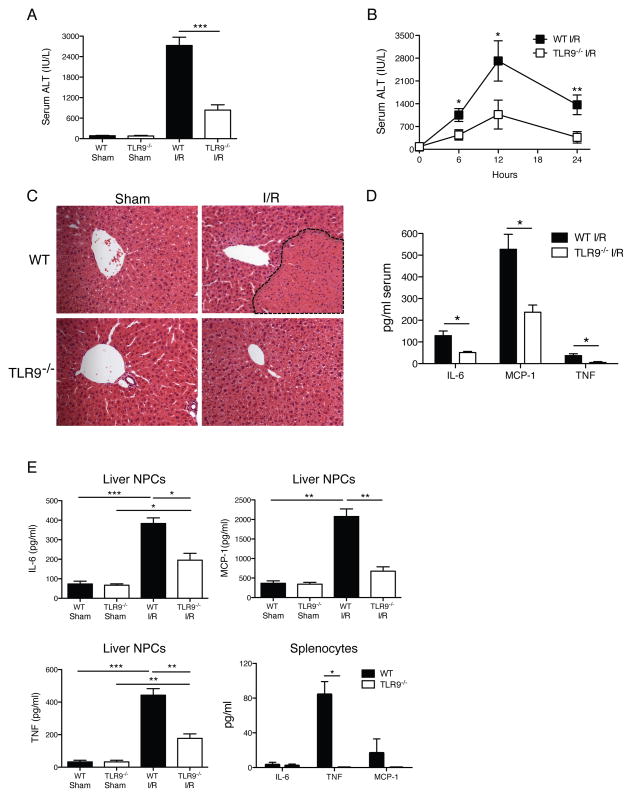
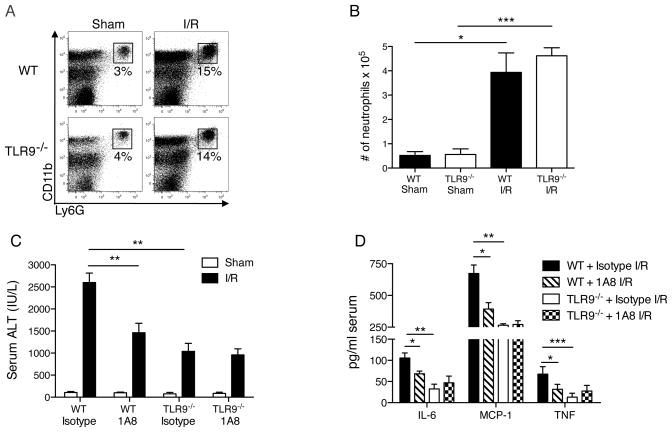
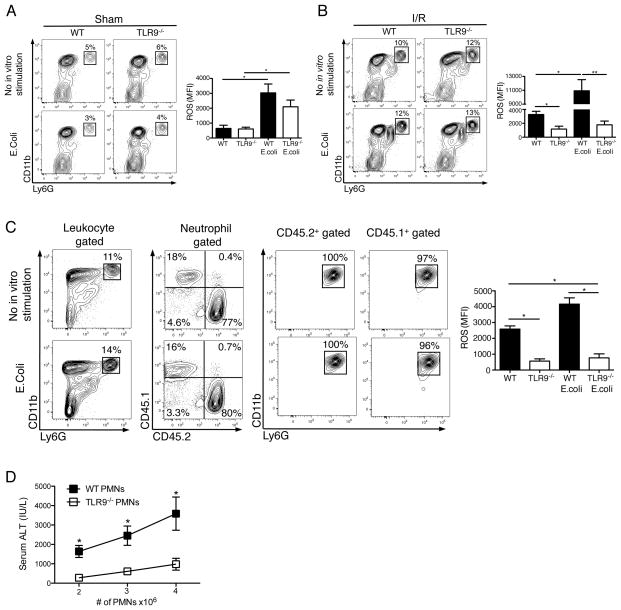
Endogenous ligands such as high-mobility group box 1 (HMGB1) and nucleic acids are released by dying cells and bind Toll-like receptors (TLRs). Because TLR9 sits at the interface of microbial and sterile inflammation by detecting both bacterial and endogenous DNA, we investigated its role in a model of segmental liver ischemia-reperfusion (I/R) injury. Mice were subjected to 1 hour of ischemia and 12 hours of reperfusion before assessment of liver injury, cytokines, and reactive oxygen species (ROS). Wild-type (WT) mice treated with an inhibitory cytosine-guanosine dinucleotide (iCpG) sequence and TLR9(-/-) mice had markedly reduced serum alanine aminotransferase (ALT) and inflammatory cytokines after liver I/R. Liver damage was mediated by bone marrow-derived cells because WT mice transplanted with TLR9(-/-) bone marrow were protected from hepatic I/R injury. Injury in WT mice partly depended on TLR9 signaling in neutrophils, which enhanced production of ROS, interleukin-6 (IL-6), and tumor necrosis factor (TNF). In vitro, DNA released from necrotic hepatocytes increased liver nonparenchymal cell (NPC) and neutrophil cytokine secretion through a TLR9-dependent mechanism. Inhibition of both TLR9 and HMGB1 caused maximal inflammatory cytokine suppression in neutrophil cultures and conferred even greater protection from I/R injury in vivo.
Conclusion: TLR9 serves as an endogenous sensor of tissue necrosis that exacerbates the innate immune response during liver I/R. Combined blockade of TLR9 and HMGB1 represents a clinically relevant, novel approach to limiting I/R injury.
Conflict of interest statement
Figures




References
-
- Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32:169–73. - PubMed
-
- Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–57. - PubMed
-
- Lentsch AB, Yoshidome H, Kato A, Warner RL, Cheadle WG, Ward PA, Edwards MJ. Requirement for interleukin-12 in the pathogenesis of warm hepatic ischemia/reperfusion injury in mice. Hepatology. 1999;30:1448–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources