

Symmetry-restrained molecular dynamics simulations improve homology models of potassium channels
- PMID: 19902533
- PMCID: PMC2811770
- DOI: 10.1002/prot.22618
Symmetry-restrained molecular dynamics simulations improve homology models of potassium channels
Abstract
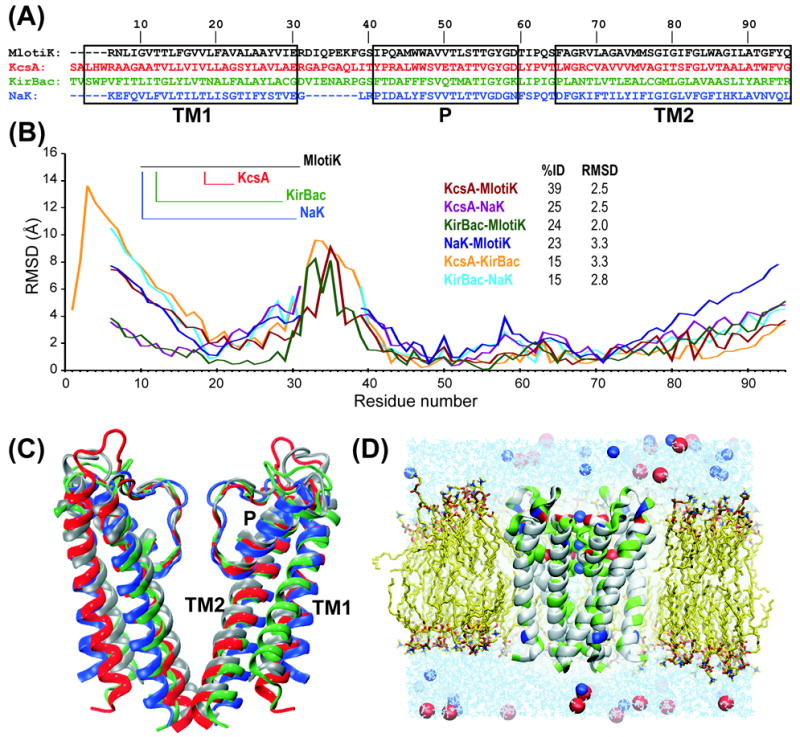
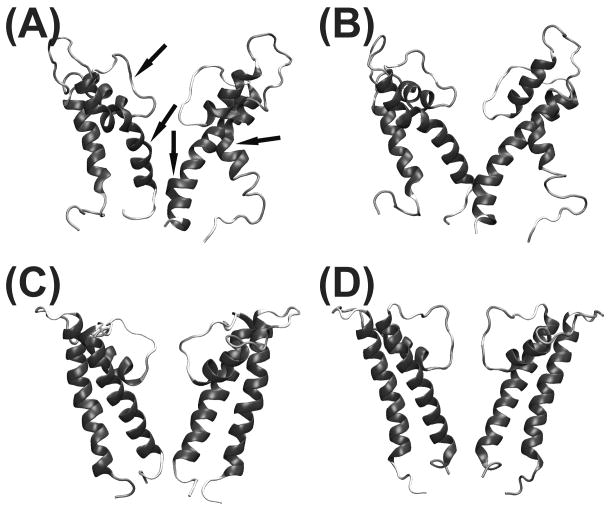
Most crystallized homo-oligomeric ion channels are highly symmetric, which dramatically decreases conformational space and facilitates building homology models (HMs). However, in molecular dynamics (MD) simulations channels deviate from ideal symmetry and accumulate thermal defects, which complicate the refinement of HMs using MD. In this work we evaluate the ability of symmetry constrained MD simulations to improve HMs accuracy, using an approach conceptually similar to Critical Assessment of techniques for protein Structure Prediction (CASP) competition: build HMs of channels with known structure and evaluate the efficiency of proposed methods in improving HMs accuracy (measured as deviation from experimental structure). Results indicate that unrestrained MD does not improve the accuracy of HMs, instantaneous symmetrization improves accuracy but not stability of HMs during subsequent unrestrained MD, while gradually imposing symmetry constraints improves both accuracy (by 5-50%) and stability of HMs. Moreover, accuracy and stability are strongly correlated, making stability a reliable criterion in predicting the accuracy of new HMs. Proteins 2010. (c) 2009 Wiley-Liss, Inc.
Figures







References
-
- Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25(10):1119–1126. - PubMed
-
- Ginalski K. Comparative modeling for protein structure prediction. Curr Opin Struct Biol. 2006;16(2):172–177. - PubMed
-
- Kryshtafovych A, Venclovas C, Fidelis K, Moult J. Progress over the first decade of CASP experiments. Proteins. 2005;61 7:225–236. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
