

Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes
- PMID: 19903739
- PMCID: PMC2809957
- DOI: 10.2337/db09-0129
Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes
Abstract
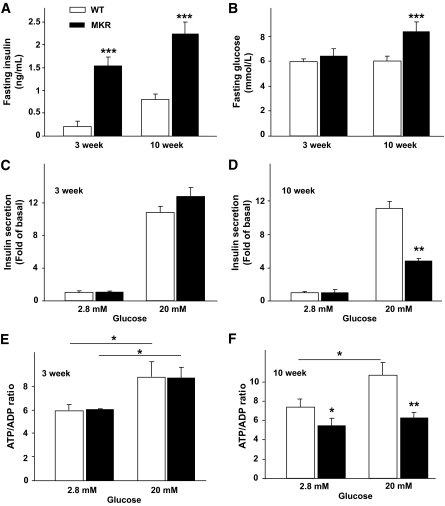
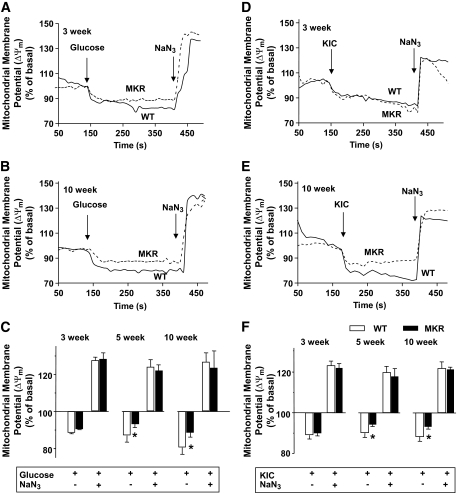
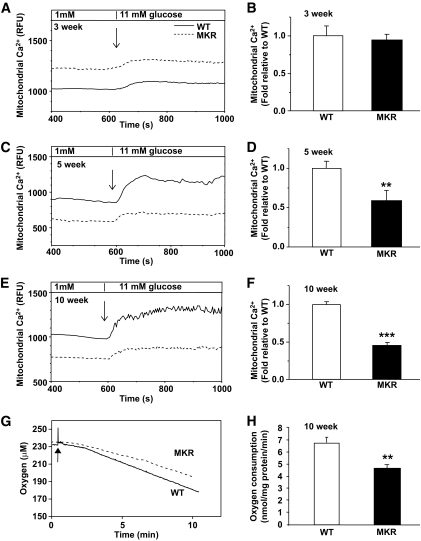
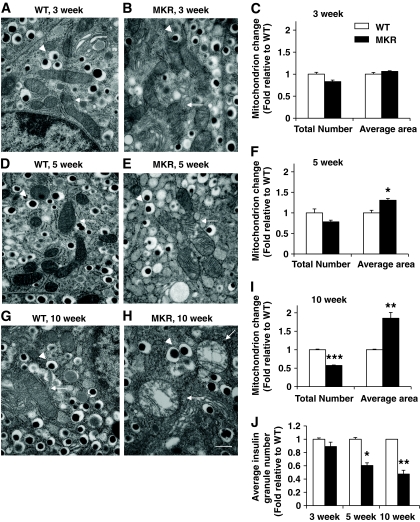
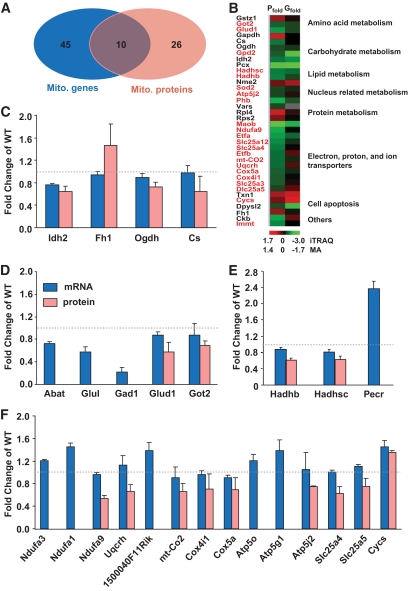
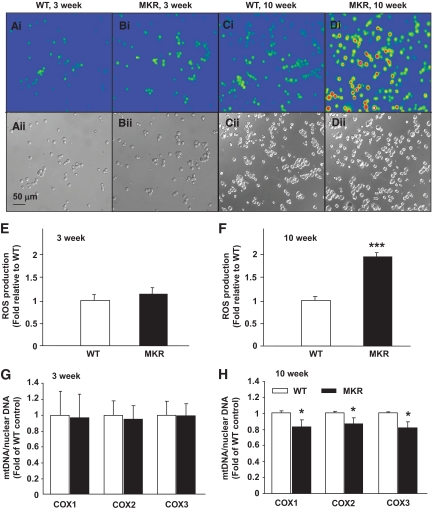
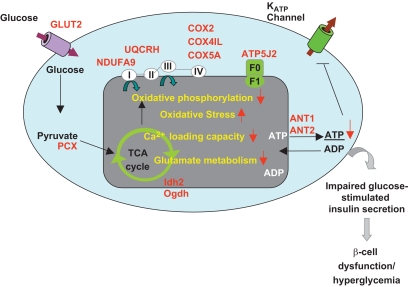
OBJECTIVE The inability of pancreatic beta-cells to appropriately respond to glucose and secrete insulin are primary defects associated with beta-cell failure in type 2 diabetes. Mitochondrial dysfunction has been implicated as a key factor in the development of type 2 diabetes; however, a link between mitochondrial dysfunction and defective insulin secretion is unclear. RESEARCH DESIGN AND METHODS We investigated the changes in islet mitochondrial function and morphology during progression from insulin resistance (3 weeks old), immediately before hyperglycemia (5 weeks old), and after diabetes onset (10 weeks old) in transgenic MKR mice compared with controls. The molecular and protein changes at 10 weeks were determined using microarray and iTRAQ proteomic screens. RESULTS At 3 weeks, MKR mice were hyperinsulinemic but normoglycemic and beta-cells showed negligible mitochondrial or morphological changes. At 5 weeks, MKR islets displayed abrogated hyperpolarization of mitochondrial membrane potential (DeltaPsi(m)), reduced mitochondrial Ca(2+) uptake, slightly enlarged mitochondria, and reduced glucose-stimulated insulin secretion. By 10 weeks, MKR mice were hyperglycemic and hyperinsulinemic and beta-cells contained swollen mitochondria with disordered cristae. beta-Cells displayed impaired stimulus-secretion coupling including reduced hyperpolarization of DeltaPsi(m), impaired Ca(2+)-signaling, and reduced glucose-stimulated ATP/ADP and insulin release. Furthermore, decreased cytochrome c oxidase-dependent oxygen consumption and signs of oxidative stress were observed in diabetic islets. Protein profiling of diabetic islets revealed that 36 mitochondrial proteins were differentially expressed, including inner membrane proteins of the electron transport chain. CONCLUSIONS We provide novel evidence for a critical role of defective mitochondrial oxidative phosphorylation and morphology in the pathology of insulin resistance-induced beta-cell failure.
Figures
References
-
- Eriksson J, Franssila-Kallunki A, Ekstrand A, Saloranta C, Widen E, Schalin C, Groop L: Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. N Engl J Med 1989;321:337–343 - PubMed
-
- Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Pettiti M, Natali A, Mari A, DeFronzo RA: Predominant role of reduced beta-cell sensitivity to glucose over insulin resistance in impaired glucose tolerance. Diabetologia 2003;46:1211–1219 - PubMed
-
- Matschinsky FM, Magnuson MA, Zelent D, Jetton TL, Doliba N, Han Y, Taub R, Grimsby J: The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes 2006;55:1–12 - PubMed
-
- MacDonald MJ, Fahien LA: Insulin release in pancreatic islets by a glycolytic and a Krebs cycle intermediate: contrasting patterns of glyceraldehyde phosphate and succinate. Arch Biochem Biophys 1990;279:104–108 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
