

Sole-Search: an integrated analysis program for peak detection and functional annotation using ChIP-seq data
- PMID: 19906703
- PMCID: PMC2817454
- DOI: 10.1093/nar/gkp1012
Sole-Search: an integrated analysis program for peak detection and functional annotation using ChIP-seq data
Abstract
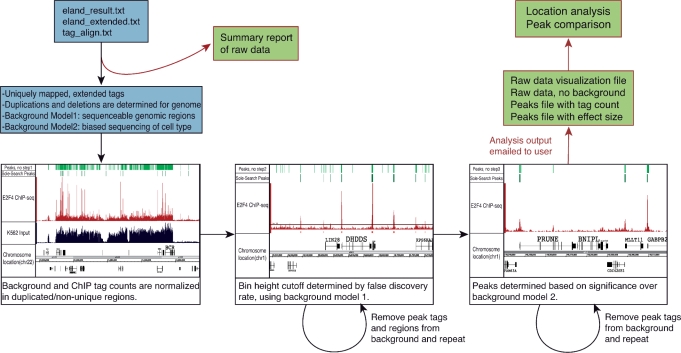
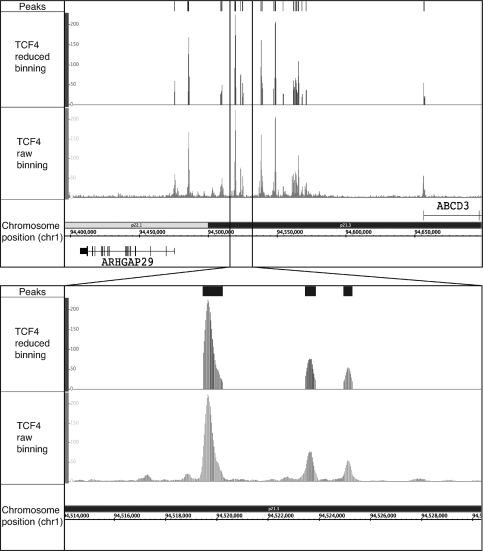
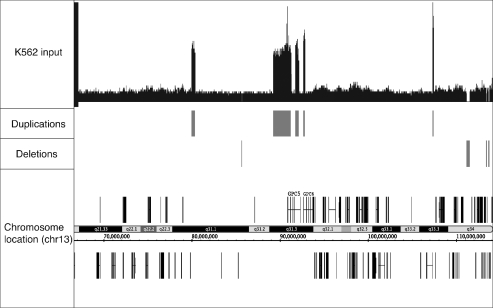
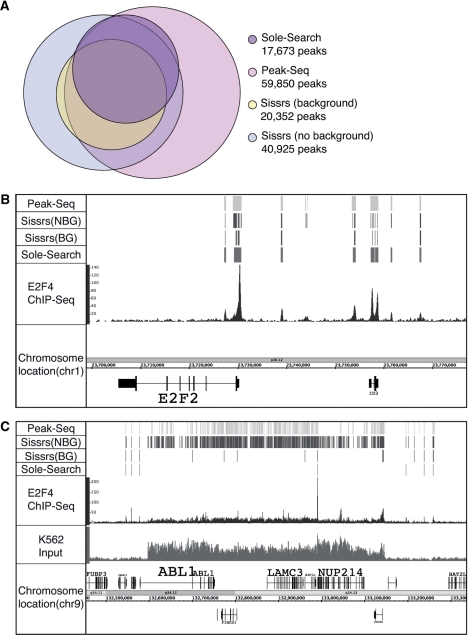
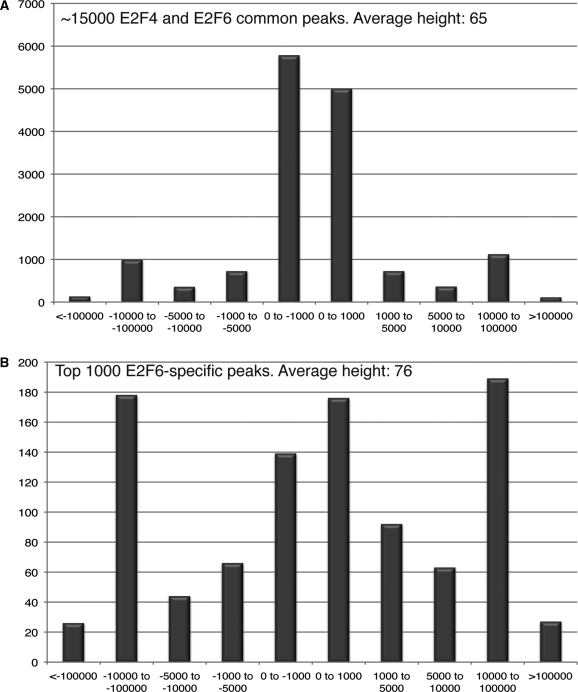
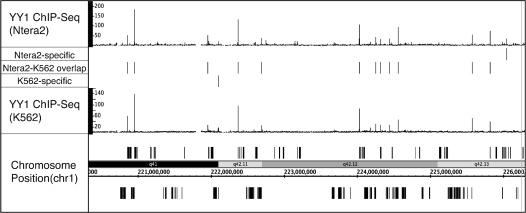
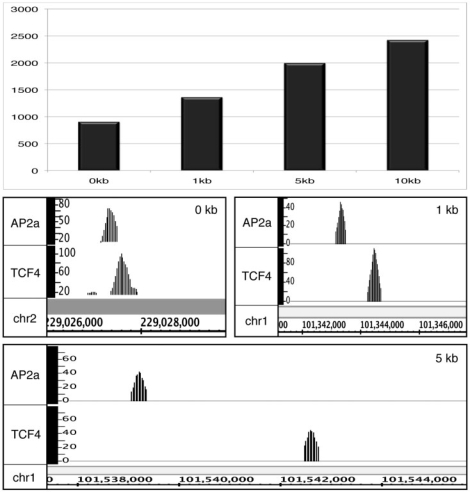
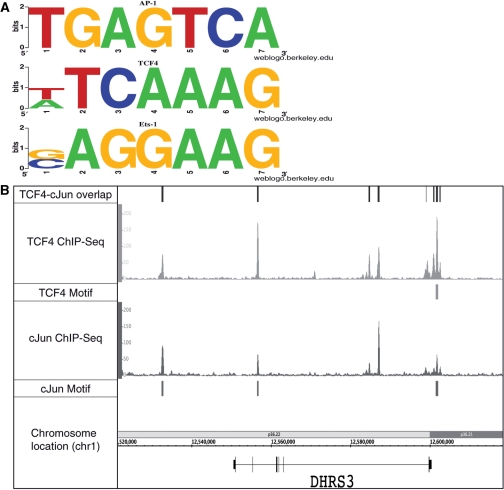
Next-generation sequencing is revolutionizing the identification of transcription factor binding sites throughout the human genome. However, the bioinformatics analysis of large datasets collected using chromatin immunoprecipitation and high-throughput sequencing is often a roadblock that impedes researchers in their attempts to gain biological insights from their experiments. We have developed integrated peak-calling and analysis software (Sole-Search) which is available through a user-friendly interface and (i) converts raw data into a format for visualization on a genome browser, (ii) outputs ranked peak locations using a statistically based method that overcomes the significant problem of false positives, (iii) identifies the gene nearest to each peak, (iv) classifies the location of each peak relative to gene structure, (v) provides information such as the number of binding sites per chromosome and per gene and (vi) allows the user to determine overlap between two different experiments. In addition, the program performs an analysis of amplified and deleted regions of the input genome. This software is web-based and automated, allowing easy and immediate access to all investigators. We demonstrate the utility of our software by collecting, analyzing and comparing ChIP-seq data for six different human transcription factors/cell line combinations.
Figures

References
-
- Oberley MJ, Inman D, Farnham PJ. E2F6 negatively regulates BRCA1 in human cancer cells without methylation of histone H3 on lysine 9. J. Biol. Chem. 2003;278:42466–42476. - PubMed
