

Genome-wide association studies in cancer--current and future directions
- PMID: 19906782
- PMCID: PMC2860704
- DOI: 10.1093/carcin/bgp273
Genome-wide association studies in cancer--current and future directions
Abstract
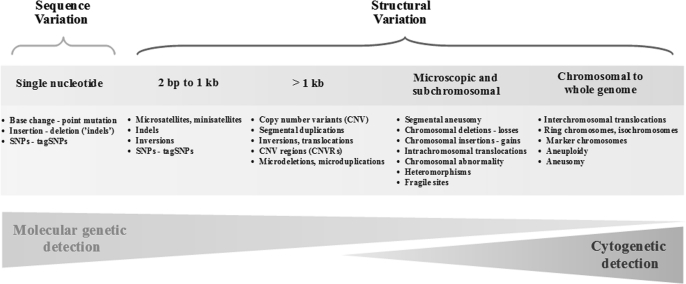
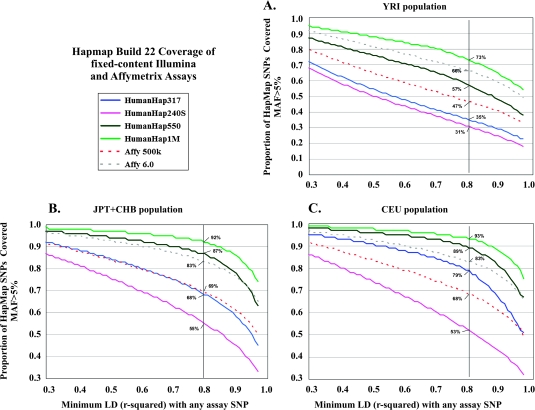
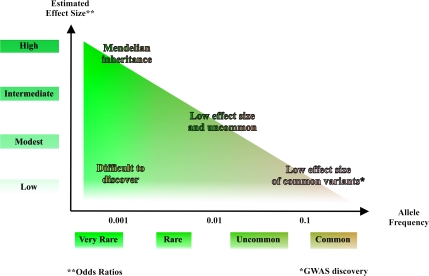
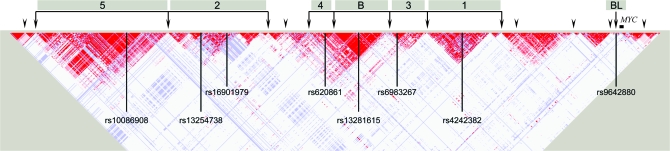
Genome-wide association studies (GWAS) have emerged as an important tool for discovering regions of the genome that harbor genetic variants that confer risk for different types of cancers. The success of GWAS in the last 3 years is due to the convergence of new technologies that can genotype hundreds of thousands of single-nucleotide polymorphism markers together with comprehensive annotation of genetic variation. This approach has provided the opportunity to scan across the genome in a sufficiently large set of cases and controls without a set of prior hypotheses in search of susceptibility alleles with low effect sizes. Generally, the susceptibility alleles discovered thus far are common, namely, with a frequency in one or more population of >10% and each allele confers a small contribution to the overall risk for the disease. For nearly all regions conclusively identified by GWAS, the per allele effect sizes estimated are <1.3. Consequently, the findings of GWAS underscore the complex nature of cancer and have focused attention on a subset of the genetic variants that comprise the genomic architecture of each type of cancer, which already can differ substantially by the number of regions associated with specific types of cancer. For instance, in prostate cancer, there could be >30 distinct regions harboring common susceptibility alleles identified by GWAS, whereas in lung cancer, a disease strongly driven by exposure to tobacco products, so far, only three regions have been conclusively established. To date, >85 regions have been conclusively associated in over a dozen different cancers, yet no more than five regions have been associated with more than one distinct cancer type. GWAS are an important discovery tool that require extensive follow-up to map each region, investigate the biological mechanism underpinning the association and eventually test the optimal markers for assessing risk for a disease or its outcome, such as in pharmacogenomics, the study of the effect of genetic variation on pharmacological interventions. The success of GWAS has opened new horizons for exploration and highlighted the complex genomic architecture of disease susceptibility.
Figures
References
-
- The International HapMap Consortium. (2003) The International HapMap Project. Nature, 426, 789–796. - PubMed
-
- Collins FS, et al. A vision for the future of genomics research. Nature. 2003;422:835–847. - PubMed
-
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–1351. - PubMed
