

Quantifying uncertainty in genotype calls
- PMID: 19906825
- PMCID: PMC2804295
- DOI: 10.1093/bioinformatics/btp624
Quantifying uncertainty in genotype calls
Abstract
Motivation: Genome-wide association studies (GWAS) are used to discover genes underlying complex, heritable disorders for which less powerful study designs have failed in the past. The number of GWAS has skyrocketed recently with findings reported in top journals and the mainstream media. Microarrays are the genotype calling technology of choice in GWAS as they permit exploration of more than a million single nucleotide polymorphisms (SNPs) simultaneously. The starting point for the statistical analyses used by GWAS to determine association between loci and disease is making genotype calls (AA, AB or BB). However, the raw data, microarray probe intensities, are heavily processed before arriving at these calls. Various sophisticated statistical procedures have been proposed for transforming raw data into genotype calls. We find that variability in microarray output quality across different SNPs, different arrays and different sample batches have substantial influence on the accuracy of genotype calls made by existing algorithms. Failure to account for these sources of variability can adversely affect the quality of findings reported by the GWAS.
Results: We developed a method based on an enhanced version of the multi-level model used by CRLMM version 1. Two key differences are that we now account for variability across batches and improve the call-specific assessment of each call. The new model permits the development of quality metrics for SNPs, samples and batches of samples. Using three independent datasets, we demonstrate that the CRLMM version 2 outperforms CRLMM version 1 and the algorithm provided by Affymetrix, Birdseed. The main advantage of the new approach is that it enables the identification of low-quality SNPs, samples and batches.
Availability: Software implementing of the method described in this article is available as free and open source code in the crlmm R/BioConductor package.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures




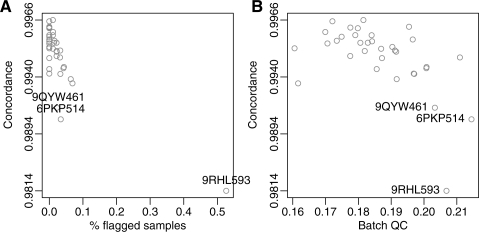
 for a given batch. Note that they are correlated. We take advantage of this correlation to predict or improve precision of shifts when not enough training data are available. The ellipses delimit the 95% confidence regions of the estimated distribution. SNPs with points outside these regions are associated with abnormal movements and are flagged as possible outliers. (A)
for a given batch. Note that they are correlated. We take advantage of this correlation to predict or improve precision of shifts when not enough training data are available. The ellipses delimit the 95% confidence regions of the estimated distribution. SNPs with points outside these regions are associated with abnormal movements and are flagged as possible outliers. (A)  versus
versus  . (B)
. (B)  versus
versus  . The plot for
. The plot for  versus
versus  is similar to that shown in (A).
is similar to that shown in (A).


References
-
- Affymetrix. Technical report. Affymetrix; 2006. BRLMM: an improved genotype calling method for the genechip human mapping 500k array set.
-
- Affymetrix. Technical report. Affymetrix; 2007. BRLMM-P: a genotype calling method for the SNP 5.0 array.
-
- Carvalho B, et al. Exploration, normalization, and genotype calls of high-density oligonucleotide SNP array data. Biostatistics. 2007;8:485–499. - PubMed
-
- Di X, et al. Dynamic model based algorithms for screening and genotyping over 100 K SNPs on oligonucleotide microarrays. Bioinformatics. 2005;21:1958–1963. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
