

NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease
- PMID: 19907078
- PMCID: PMC2786797
- DOI: 10.1172/JCI39716
NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease
Abstract
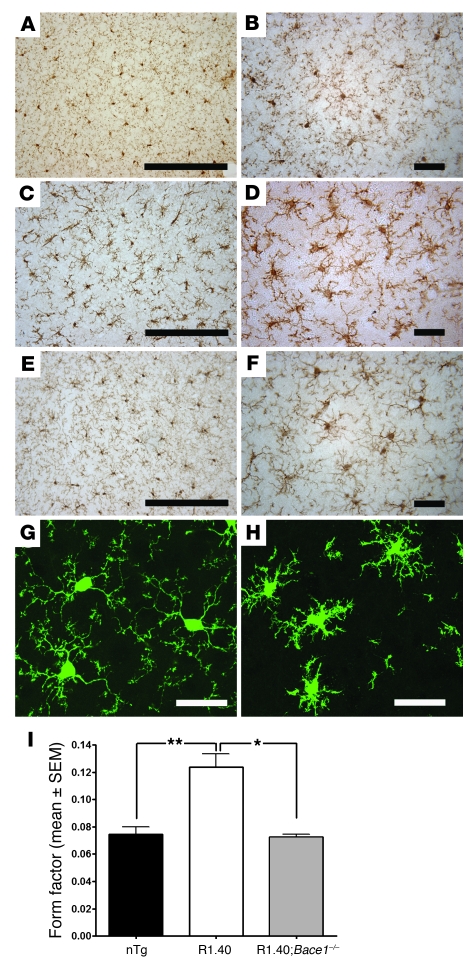
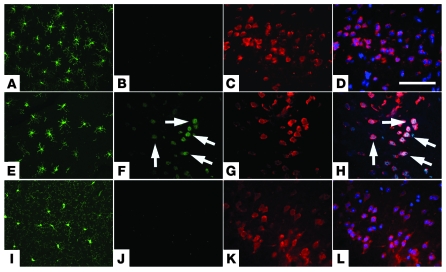
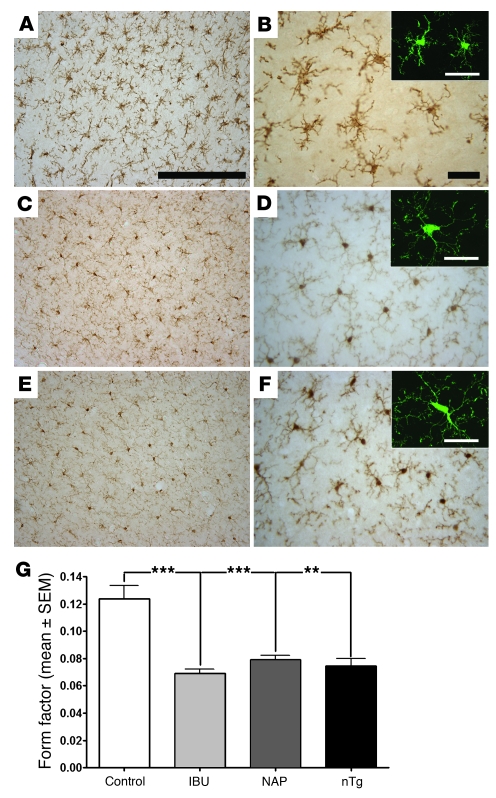
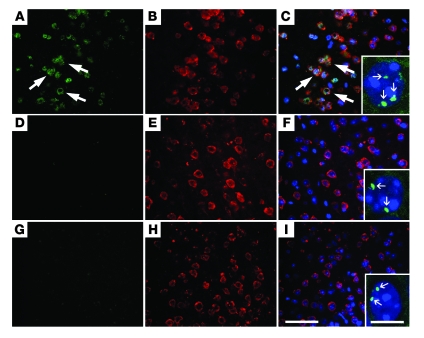
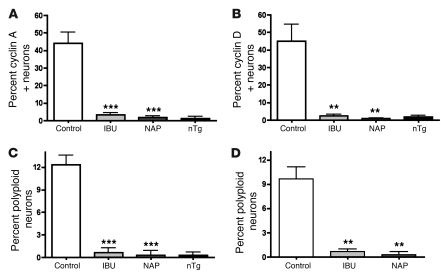
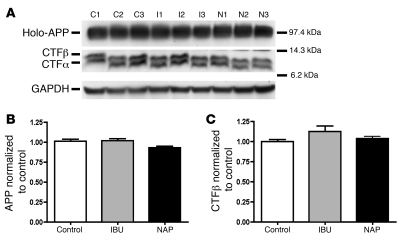
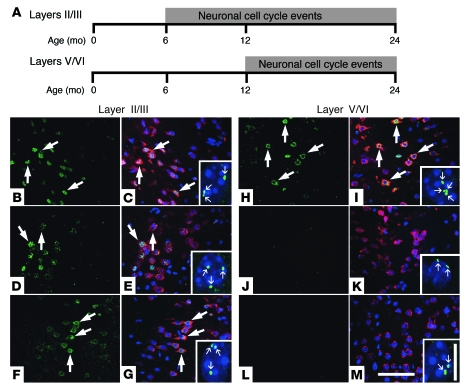
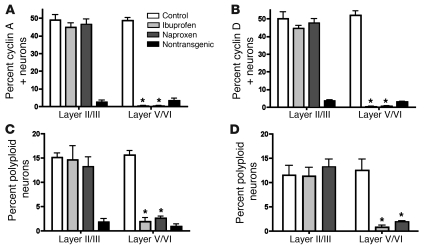
Ectopic cell cycle events (CCEs) mark vulnerable neuronal populations in human Alzheimer disease (AD) and are observed early in disease progression. In transgenic mouse models of AD, CCEs are found before the onset of beta-amyloid peptide (Abeta) deposition to form senile plaques, a hallmark of AD. Here, we have demonstrated that alterations in brain microglia occur coincidently with the appearance of CCEs in the R1.40 transgenic mouse model of AD. Furthermore, promotion of inflammation with LPS at young ages in R1.40 mice induced the early appearance of neuronal CCEs, whereas treatment with 2 different nonsteroidal antiinflammatory drugs (NSAIDs) blocked neuronal CCEs and alterations in brain microglia without altering amyloid precursor protein (APP) processing and steady-state Abeta levels. In addition, NSAID treatment of older R1.40 animals prevented new neuronal CCEs, although it failed to reverse existing ones. Retrospective human epidemiological studies have identified long-term use of NSAIDs as protective against AD. Prospective clinical trials, however, have failed to demonstrate a similar benefit. Our use of CCEs as an outcome measure offers fresh insight into this discrepancy and provides important information for future clinical trials, as it suggests that NSAID use in human AD may need to be initiated as early as possible to prevent disease progression.
Figures
References
-
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 2006;12:1005–1015. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
