

Pathogenic mechanisms of myotonic dystrophy
- PMID: 19909263
- PMCID: PMC3873089
- DOI: 10.1042/BST0371281
Pathogenic mechanisms of myotonic dystrophy
Abstract
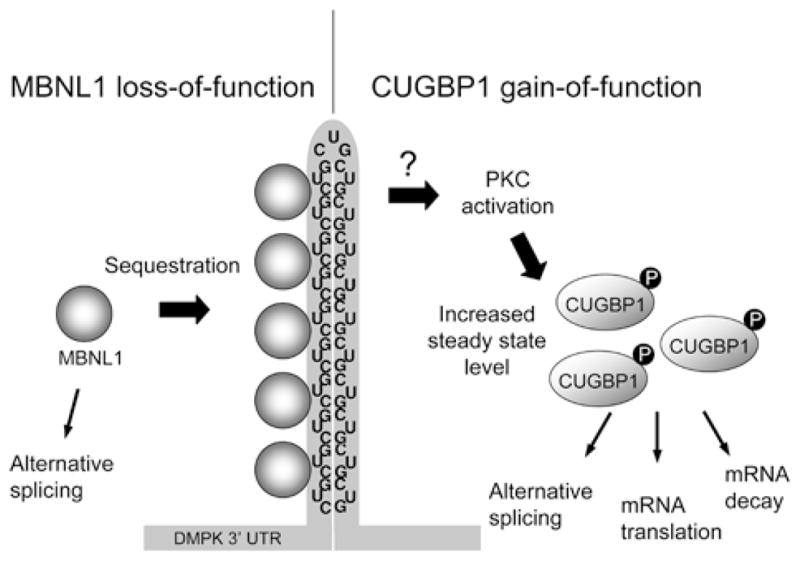
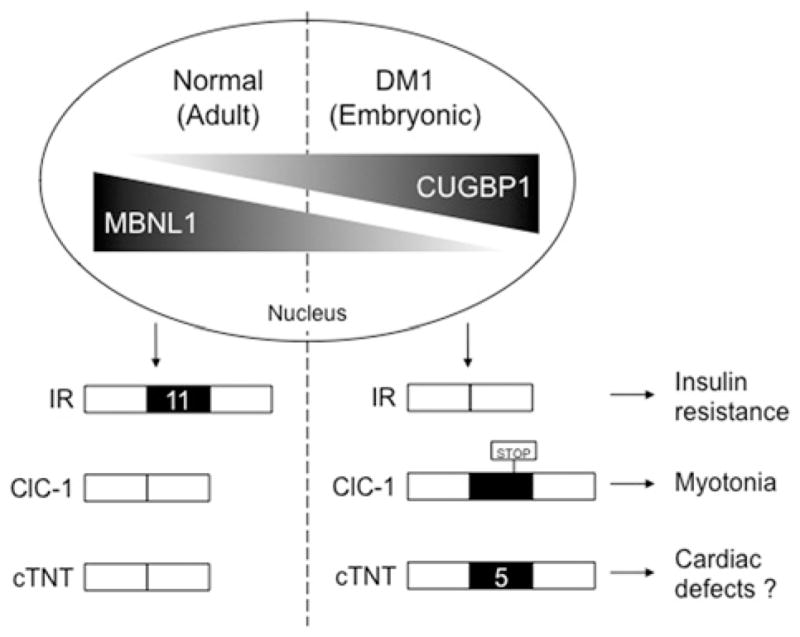
DM (myotonic dystrophy) is a dominantly inherited genetic disorder that is the most common cause of muscular dystrophy in adults affecting 1 in 8500 individuals worldwide. Different microsatellite expansions in two loci cause different forms of the disease that share similar features: DM1 (DM type 1) is caused by a tri- (CTG) nucleotide expansion within the DMPK (dystrophia myotonica protein kinase) 3'-untranslated region and DM2 (DM type 2) is caused by a tetra- (CCTG) nucleotide expansion within intron 1 of the ZNF9 (zinc finger 9) gene. The pathogenic mechanism of this disease involves the RNA transcribed from the expanded allele containing long tracts of (CUG)(n) or (CCUG)(n). The RNA results in a toxic effect through two RNA-binding proteins: MBNL1 (muscleblind-like 1) and CUGBP1 (CUG-binding protein 1). In DM1, MBNL1 is sequestered on CUG repeat-containing RNA resulting in its loss-of-function, while CUGBP1 is up-regulated through a signalling pathway. The downstream effects include disrupted regulation of alternative splicing, mRNA translation and mRNA stability, which contribute to the multiple features of DM1. This review will focus on the RNA gain-of-function disease mechanism, the important roles of MBNL1 and CUGBP1 in DM1, and the relevance to other RNA dominant disorders.
Figures
References
-
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. - PubMed
-
- Harper P. Myotonic Dystrophy. W.B, Saunders; London: 2001.
-
- Fu YH, Friedman DL, Richards S, Pearlman JA, Gibbs RA, Pizzuti A, Ashizawa T, Perryman MB, Scarlato G, Fenwick RG, Caskey CT. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science. 1993;260:235–238. - PubMed
-
- Jansen G, Groenen P, Bachner D, Jap PHK, Coerwinkel M, Oerlemans F, vandenBroek W, Gohlsch B, Pette D, Plomp JJ, et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet. 1996;13:316–324. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous