

Composite likelihood estimation of demographic parameters
- PMID: 19909534
- PMCID: PMC2783031
- DOI: 10.1186/1471-2156-10-72
Composite likelihood estimation of demographic parameters
Abstract
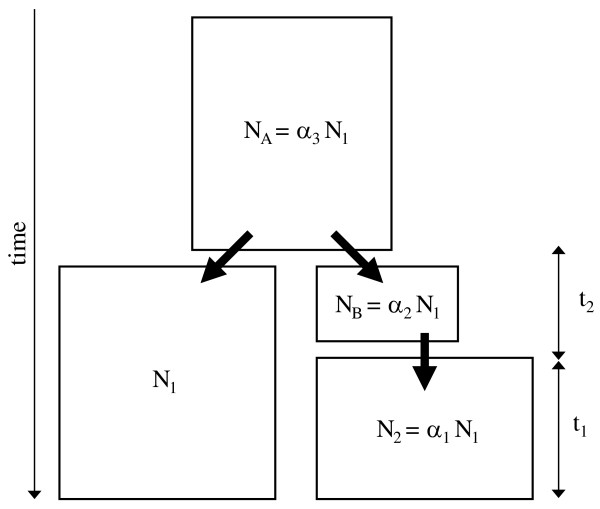
Background: Most existing likelihood-based methods for fitting historical demographic models to DNA sequence polymorphism data to do not scale feasibly up to the level of whole-genome data sets. Computational economies can be achieved by incorporating two forms of pseudo-likelihood: composite and approximate likelihood methods. Composite likelihood enables scaling up to large data sets because it takes the product of marginal likelihoods as an estimator of the likelihood of the complete data set. This approach is especially useful when a large number of genomic regions constitutes the data set. Additionally, approximate likelihood methods can reduce the dimensionality of the data by summarizing the information in the original data by either a sufficient statistic, or a set of statistics. Both composite and approximate likelihood methods hold promise for analyzing large data sets or for use in situations where the underlying demographic model is complex and has many parameters. This paper considers a simple demographic model of allopatric divergence between two populations, in which one of the population is hypothesized to have experienced a founder event, or population bottleneck. A large resequencing data set from human populations is summarized by the joint frequency spectrum, which is a matrix of the genomic frequency spectrum of derived base frequencies in two populations. A Bayesian Metropolis-coupled Markov chain Monte Carlo (MCMCMC) method for parameter estimation is developed that uses both composite and likelihood methods and is applied to the three different pairwise combinations of the human population resequence data. The accuracy of the method is also tested on data sets sampled from a simulated population model with known parameters.
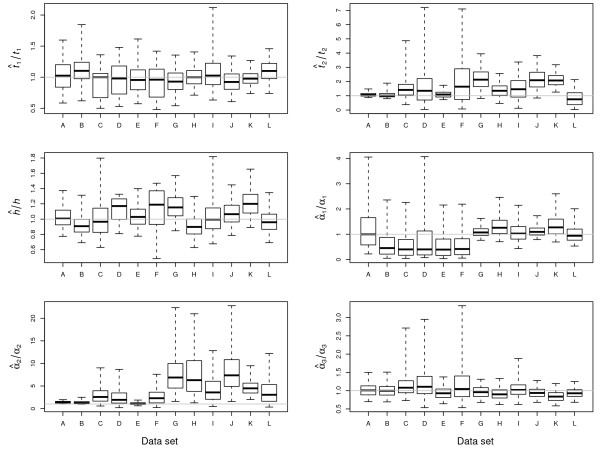
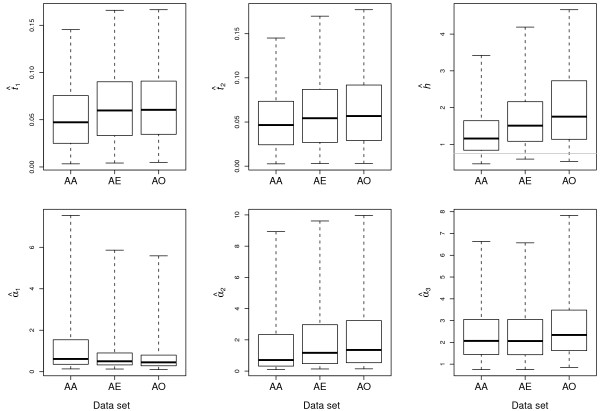
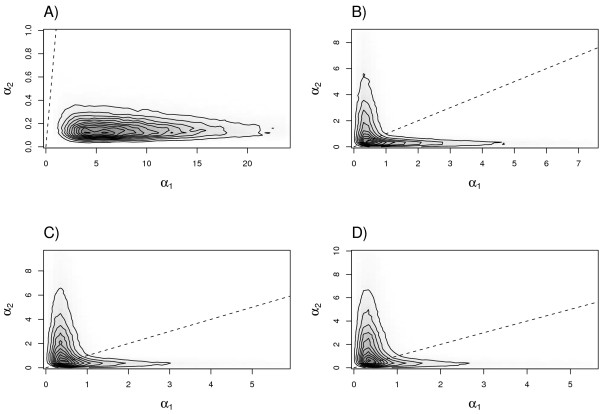
Results: The Bayesian MCMCMC method also estimates the ratio of effective population size for the X chromosome versus that of the autosomes. The method is shown to estimate, with reasonable accuracy, demographic parameters from three simulated data sets that vary in the magnitude of a founder event and a skew in the effective population size of the X chromosome relative to the autosomes. The behavior of the Markov chain is also examined and shown to convergence to its stationary distribution, while also showing high levels of parameter mixing. The analysis of three pairwise comparisons of sub-Saharan African human populations with non-African human populations do not provide unequivocal support for a strong non-African founder event from these nuclear data. The estimates do however suggest a skew in the ratio of X chromosome to autosome effective population size that is greater than one. However in all three cases, the 95% highest posterior density interval for this ratio does include three-fourths, the value expected under an equal breeding sex ratio.
Conclusion: The implementation of composite and approximate likelihood methods in a framework that includes MCMCMC demographic parameter estimation shows great promise for being flexible and computationally efficient enough to scale up to the level of whole-genome polymorphism and divergence analysis. Further work must be done to characterize the effects of the assumption of linkage equilibrium among genomic regions that is crucial to the validity of applying the composite likelihood method.
Figures
References
-
- Lindsay BG. Composite likelihood methods. Contemp Math. 1988;80:221–239.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
