

Cisplatin enhances protein kinase R-like endoplasmic reticulum kinase- and CD95-dependent melanoma differentiation-associated gene-7/interleukin-24-induced killing in ovarian carcinoma cells
- PMID: 19910452
- PMCID: PMC2812067
- DOI: 10.1124/mol.109.061820
Cisplatin enhances protein kinase R-like endoplasmic reticulum kinase- and CD95-dependent melanoma differentiation-associated gene-7/interleukin-24-induced killing in ovarian carcinoma cells
Abstract
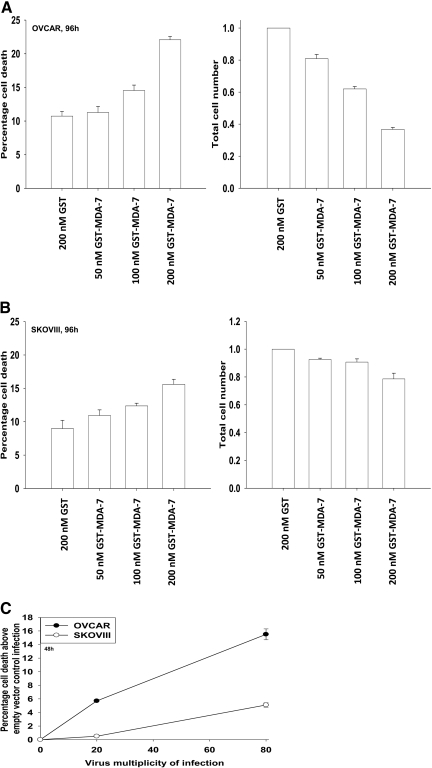
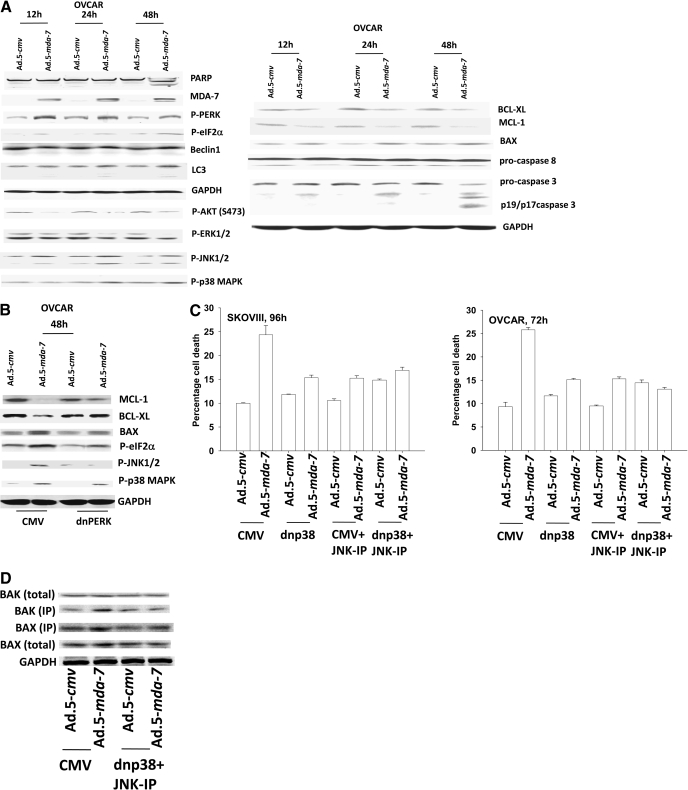
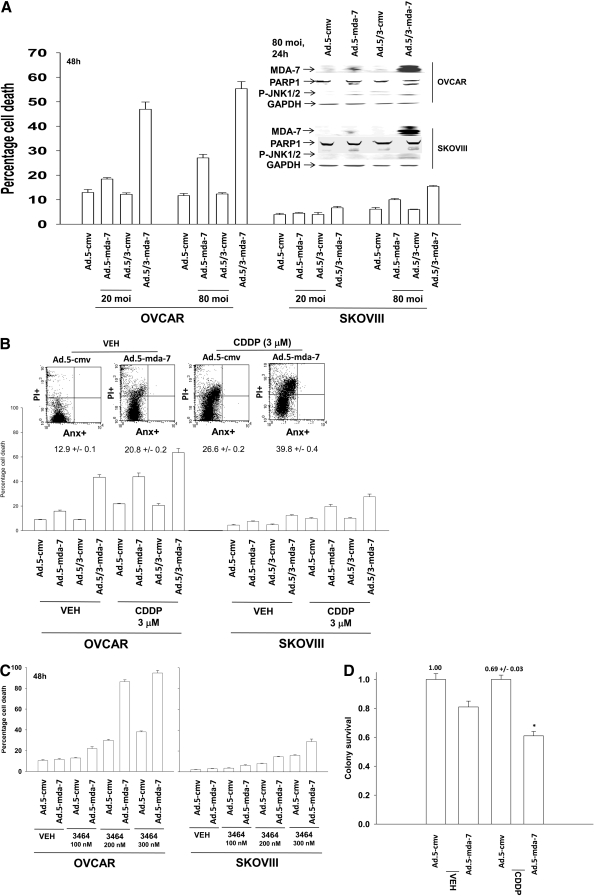
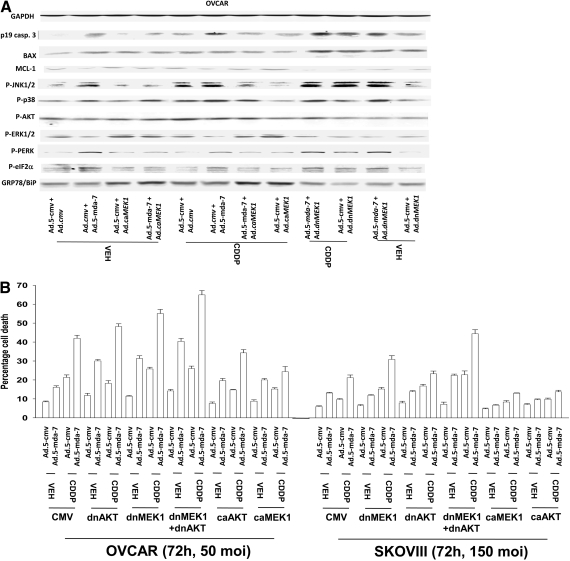
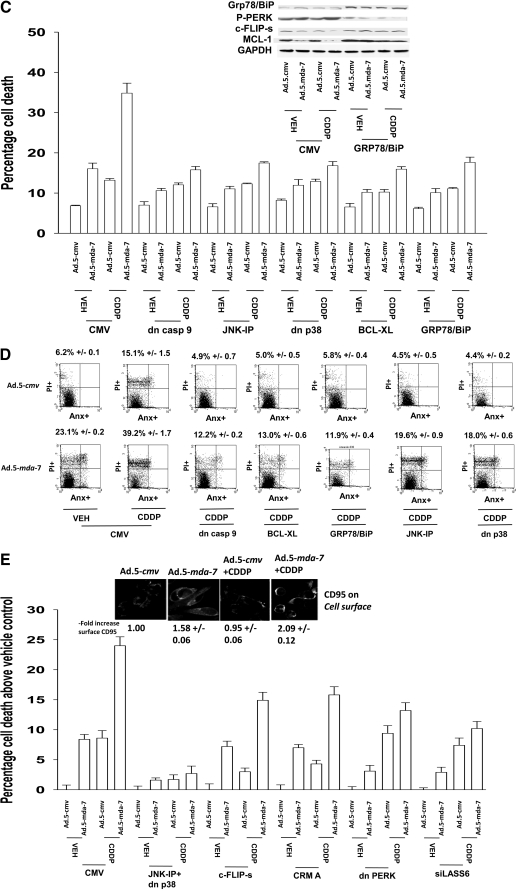
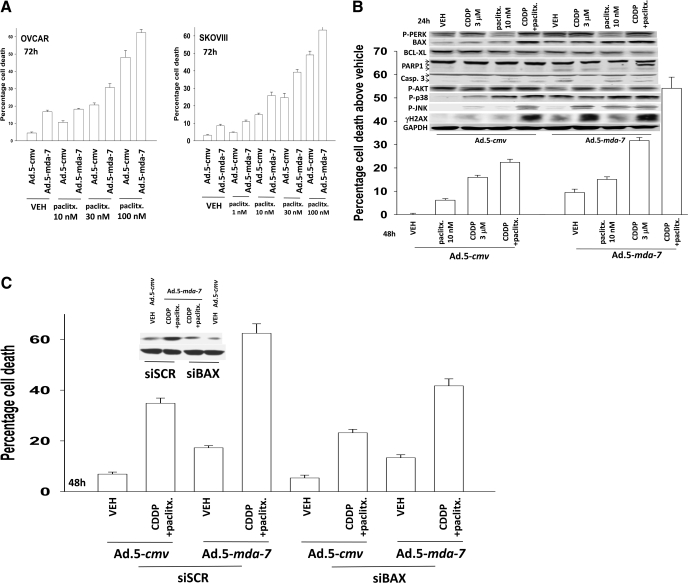
Melanoma differentiation associated gene-7/interleukin 24 (mda-7/IL-24) is a unique interleukin (IL)-10 family cytokine displaying selective apoptosis-inducing activity in transformed cells without harming normal cells. The present studies focused on defining the mechanism(s) by which recombinant adenoviral delivery of MDA-7/IL-24 inhibits cell survival of human ovarian carcinoma cells. Expression of MDA-7/IL-24 induced phosphorylation of protein kinase R-like endoplasmic reticulum kinase (PERK) and eukaryotic initiation factor2alpha (eIF2alpha). In a PERK-dependent fashion, MDA-7/IL-24 reduced ERK1/2 and AKT phosphorylation and activated c-Jun NH(2)-terminal kinase (JNK) 1/2 and p38 mitogen-activated protein kinase (MAPK). MDA-7/IL-24 reduced MCL-1 and BCL-XL and increased BAX levels via PERK signaling; cell-killing was mediated via the intrinsic pathway, and cell killing was primarily necrotic as judged using Annexin V/propidium iodide staining. Inhibition of p38 MAPK and JNK1/2 abolished MDA-7/IL-24 toxicity and blocked BAX and BAK activation, whereas activation of mitogen-activated extracellular-regulated kinase (MEK) 1/2 or AKT suppressed enhanced killing and JNK1/2 activation. MEK1/2 signaling increased expression of the MDA-7/IL-24 and PERK chaperone BiP/78-kDa glucose regulated protein (GRP78), and overexpression of BiP/GRP78 suppressed MDA-7/IL-24 toxicity. MDA-7/IL-24-induced LC3-green fluorescent protein vesicularization and processing of LC3; and knockdown of ATG5 suppressed MDA-7/IL-24-mediated toxicity. MDA-7/IL-24 and cisplatin interacted in a greater than additive fashion to kill tumor cells that was dependent on a further elevation of JNK1/2 activity and recruitment of the extrinsic CD95 pathway. MDA-7/IL-24 toxicity was enhanced in a weak additive fashion by paclitaxel; paclitaxel enhanced MDA-7/IL-24 + cisplatin lethality in a greater than additive fashion via BAX. Collectively, our data demonstrate that MDA-7/IL-24 induces an endoplasmic reticulum stress response that activates multiple proapoptotic pathways, culminating in decreased ovarian tumor cell survival.
Figures

Similar articles
-
MDA-7/IL-24-induced cell killing in malignant renal carcinoma cells occurs by a ceramide/CD95/PERK-dependent mechanism.Mol Cancer Ther. 2009 May;8(5):1280-91. doi: 10.1158/1535-7163.MCT-09-0073. Epub 2009 May 5. Mol Cancer Ther. 2009. PMID: 19417161 Free PMC article.
-
Caspase-, cathepsin-, and PERK-dependent regulation of MDA-7/IL-24-induced cell killing in primary human glioma cells.Mol Cancer Ther. 2008 Feb;7(2):297-313. doi: 10.1158/1535-7163.MCT-07-2166. Mol Cancer Ther. 2008. PMID: 18281515 Free PMC article.
-
PERK-dependent regulation of MDA-7/IL-24-induced autophagy in primary human glioma cells.Autophagy. 2008 May;4(4):513-5. doi: 10.4161/auto.5725. Epub 2008 Feb 13. Autophagy. 2008. PMID: 18299661 Free PMC article.
-
Pathogenicity and virulence of Japanese encephalitis virus: Neuroinflammation and neuronal cell damage.Virulence. 2021 Dec;12(1):968-980. doi: 10.1080/21505594.2021.1899674. Virulence. 2021. PMID: 33724154 Free PMC article. Review.
-
Endoplasmic reticulum stress in liver disease.J Hepatol. 2011 Apr;54(4):795-809. doi: 10.1016/j.jhep.2010.11.005. Epub 2010 Nov 13. J Hepatol. 2011. PMID: 21145844 Free PMC article. Review.
Cited by
-
Combinatorial strategies based on CRAd-IL24 and CRAd-ING4 virotherapy with anti-angiogenesis treatment for ovarian cancer.J Ovarian Res. 2016 Jun 27;9(1):38. doi: 10.1186/s13048-016-0248-5. J Ovarian Res. 2016. PMID: 27349517 Free PMC article.
-
Fanning the Flames of Endoplasmic Reticulum (ER) Stress: Can Sphingolipid Metabolism Be Targeted to Enhance ER Stress-Associated Immunogenic Cell Death in Cancer?Mol Pharmacol. 2024 Feb 15;105(3):155-165. doi: 10.1124/molpharm.123.000786. Mol Pharmacol. 2024. PMID: 38164594 Free PMC article. Review.
-
The development of MDA-7/IL-24 as a cancer therapeutic.Pharmacol Ther. 2010 Nov;128(2):375-84. doi: 10.1016/j.pharmthera.2010.08.001. Epub 2010 Aug 21. Pharmacol Ther. 2010. PMID: 20732354 Free PMC article. Review.
-
Histone deacetylase inhibitors interact with melanoma differentiation associated-7/interleukin-24 to kill primary human glioblastoma cells.Mol Pharmacol. 2013 Aug;84(2):171-81. doi: 10.1124/mol.113.086553. Epub 2013 May 9. Mol Pharmacol. 2013. PMID: 23661648 Free PMC article.
-
Enhanced delivery of mda-7/IL-24 using a serotype chimeric adenovirus (Ad.5/3) in combination with the Apogossypol derivative BI-97C1 (Sabutoclax) improves therapeutic efficacy in low CAR colorectal cancer cells.J Cell Physiol. 2012 May;227(5):2145-53. doi: 10.1002/jcp.22947. J Cell Physiol. 2012. PMID: 21780116 Free PMC article.
References
-
- Caudell EG, Mumm JB, Poindexter N, Ekmekcioglu S, Mhashilkar AM, Yang XH, Retter MW, Hill P, Chada S, Grimm EA. (2002) The protein product of the tumor suppressor gene, melanoma differentiation-associated gene 7, exhibits immunostimulatory activity and is designated IL-24. J Immunol 168: 6041–6046 - PubMed
-
- Chada S, Bocangel D, Ramesh R, Grimm EA, Mumm JB, Mhashilkar AM, Zheng M. (2005) mda-7/IL24 kills pancreatic cancer cells by inhibition of the Wnt/PI3K signaling pathways: identification of IL-20 receptor-mediated bystander activity against pancreatic cancer. Mol Ther 11: 724–733 - PubMed
-
- Coleman RL, Sood AK. (2006) Historical progress in the initial management of ovarian cancer: intraperitoneal chemotherapy. Curr Oncol Rep 8: 455–464 - PubMed
-
- Cunningham CC, Chada S, Merritt JA, Tong A, Senzer N, Zhang Y, Mhashilkar A, Parker K, Vukelja S, Richards D, et al. (2005) Clinical and local biological effects of an intratumoral injection of mda-7 (IL24; INGN 241) in patients with advanced carcinoma: a phase I study. Mol Ther 11: 149–159 - PubMed
-
- Dash R, Dmitriev IP, Su ZZ, Bhutia SJ, Azab B, Vozhilla N, Yacoub A, Dent P, Curiel DT, Sarkar D, et al. (2010) Enhanced delivery of mda-7/IL-24 using a serotype chimeric adenovirus (Ad.5/3) improves therapeutic efficacy in low CAR prostate cancer cells. Cancer Gene Ther, in press - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K22 CA122828/CA/NCI NIH HHS/United States
- P01-NS031492/NS/NINDS NIH HHS/United States
- R01-CA63753/CA/NCI NIH HHS/United States
- R01 CA063753/CA/NCI NIH HHS/United States
- R01-DK52825/DK/NIDDK NIH HHS/United States
- R01 CA097318/CA/NCI NIH HHS/United States
- R01-CA108325/CA/NCI NIH HHS/United States
- R01-CA12764101/CA/NCI NIH HHS/United States
- P01 CA104177/CA/NCI NIH HHS/United States
- R01 CA078754/CA/NCI NIH HHS/United States
- R01-CA77141/CA/NCI NIH HHS/United States
- P01-CA104177/CA/NCI NIH HHS/United States
- P01 NS031492/NS/NINDS NIH HHS/United States
- R01 DK052825/DK/NIDDK NIH HHS/United States
- R01-CA097318/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
