

Ethanol enhances hepatitis C virus replication through lipid metabolism and elevated NADH/NAD+
- PMID: 19910460
- PMCID: PMC2801286
- DOI: 10.1074/jbc.M109.045740
Ethanol enhances hepatitis C virus replication through lipid metabolism and elevated NADH/NAD+
Abstract
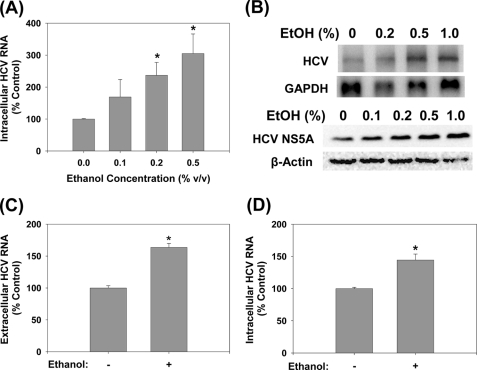
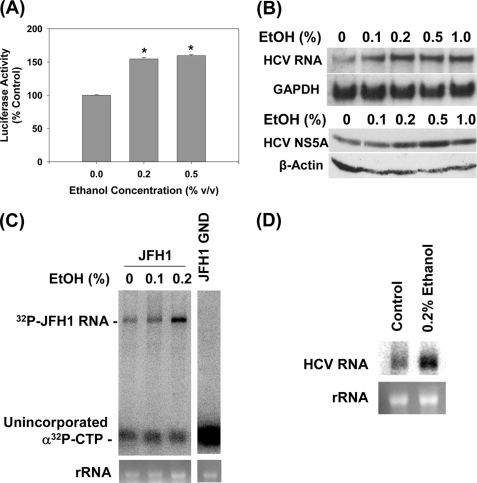
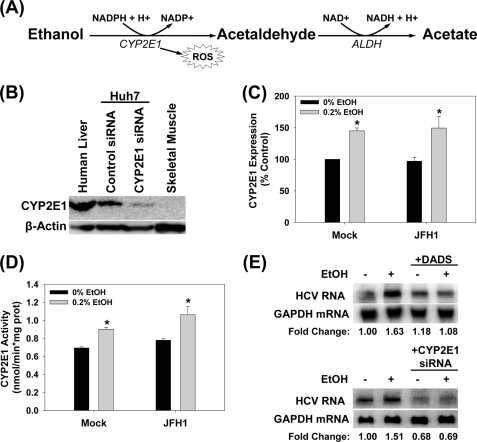
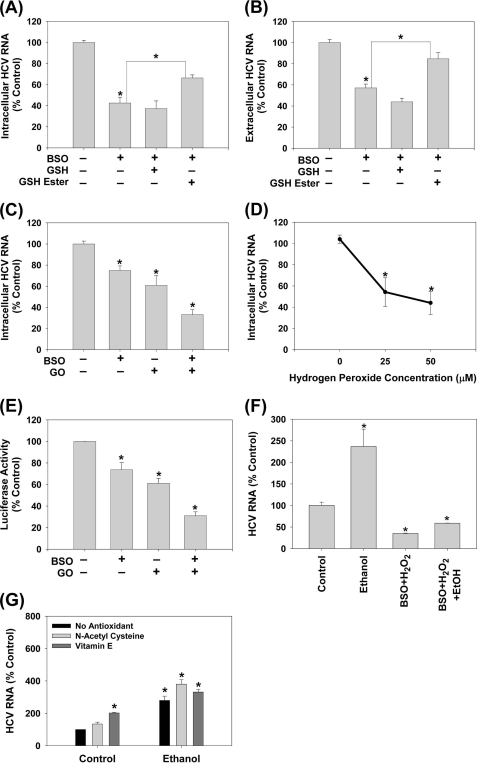
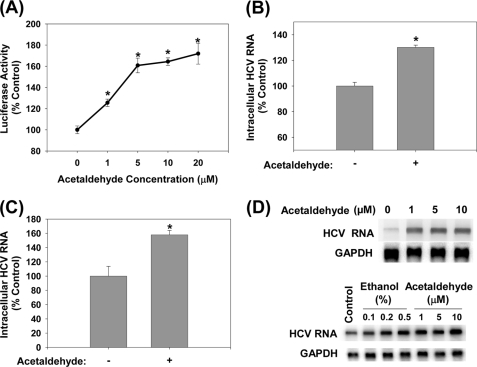
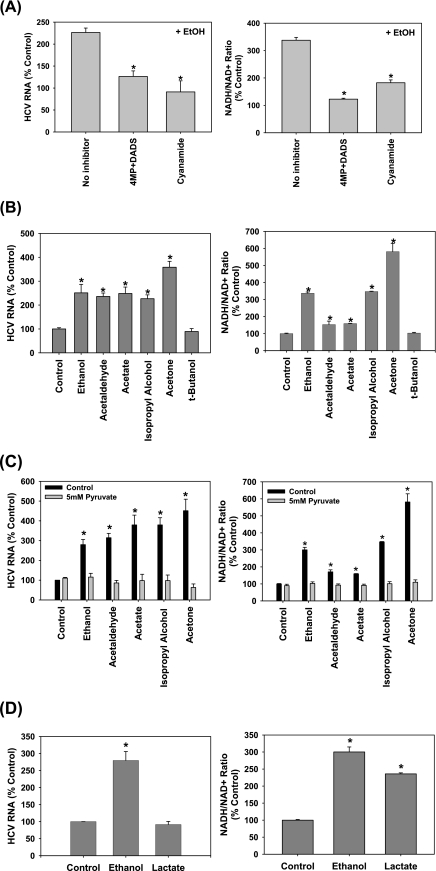
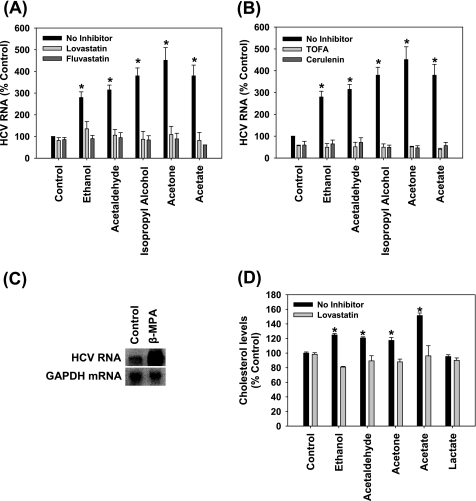
Ethanol has been suggested to elevate HCV titer in patients and to increase HCV RNA in replicon cells, suggesting that HCV replication is increased in the presence and absence of the complete viral replication cycle, but the mechanisms remain unclear. In this study, we use Huh7 human hepatoma cells that naturally express comparable levels of CYP2E1 as human liver to demonstrate that ethanol, at subtoxic and physiologically relevant concentrations, enhances complete HCV replication. The viral RNA genome replication is affected for both genotypes 2a and 1b. Acetaldehyde, a major product of ethanol metabolism, likewise enhances HCV replication at physiological concentrations. The potentiation of HCV replication by ethanol is suppressed by inhibiting CYP2E1 or aldehyde dehydrogenase and requires an elevated NADH/NAD(+) ratio. In addition, acetate, isopropyl alcohol, and concentrations of acetone that occur in diabetics enhance HCV replication with corresponding increases in the NADH/NAD(+). Furthermore, inhibiting the host mevalonate pathway with lovastatin or fluvastatin and fatty acid synthesis with 5-(tetradecyloxy)-2-furoic acid or cerulenin significantly attenuates the enhancement of HCV replication by ethanol, acetaldehyde, acetone, as well as acetate, whereas inhibiting beta-oxidation with beta-mercaptopropionic acid increases HCV replication. Ethanol, acetaldehyde, acetone, and acetate increase the total intracellular cholesterol content, which is attenuated with lovastatin. In contrast, both endogenous and exogenous ROS suppress the replication of HCV genotype 2a, as previously shown with genotype 1b.
Conclusion: Therefore, lipid metabolism and alteration of cellular NADH/NAD(+) ratio are likely to play a critical role in the potentiation of HCV replication by ethanol rather than oxidative stress.
Figures
References
-
- Jamal M. M., Morgan T. R. (2003) Best Pract. Res. Clin. Gastroenterol. 17, 649–662 - PubMed
-
- Sata M., Fukuizumi K., Uchimura Y., Nakano H., Ishii K., Kumashiro R., Mizokami M., Lau J. Y., Tanikawa K. (1996) J. Viral. Hepat. 3, 143–148 - PubMed
-
- Zhang T., Li Y., Lai J. P., Douglas S. D., Metzger D. S., O'Brien C. P., Ho W. Z. (2003) Hepatology 38, 57–65 - PubMed
-
- Seronello S., Sheikh M. Y., Choi J. (2007) Free Radic. Biol. Med. 43, 869–882 - PubMed
-
- Cromie S. L., Jenkins P. J., Bowden D. S., Dudley F. J. (1996) J. Hepatol. 25, 821–826 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
