

New hypotheses and opportunities in endocrine therapy: amplification of oestrogen-induced apoptosis
- PMID: 19914527
- PMCID: PMC2867601
- DOI: 10.1016/S0960-9776(09)70266-8
New hypotheses and opportunities in endocrine therapy: amplification of oestrogen-induced apoptosis
Abstract
Aims: To outline the progress being made in the understanding of acquired resistance to long term therapy with the selective oestrogen receptor modulators (SERMs, tamoxifen and raloxifene) and aromatase inhibitors. The question to be addressed is how we can amplify the new biology of oestrogen-induced apoptosis to create more complete responses in exhaustively antihormone treated metastatic breast cancer.
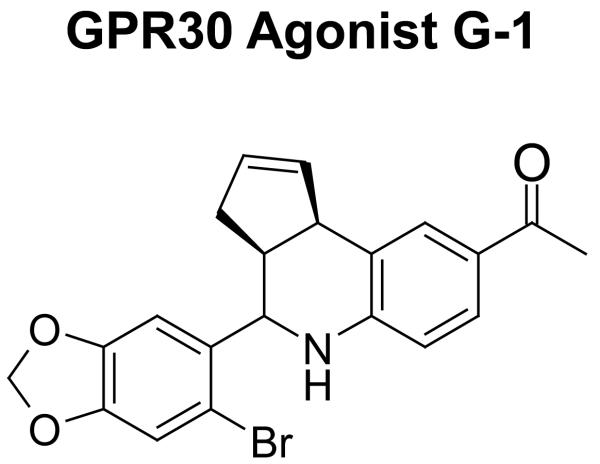
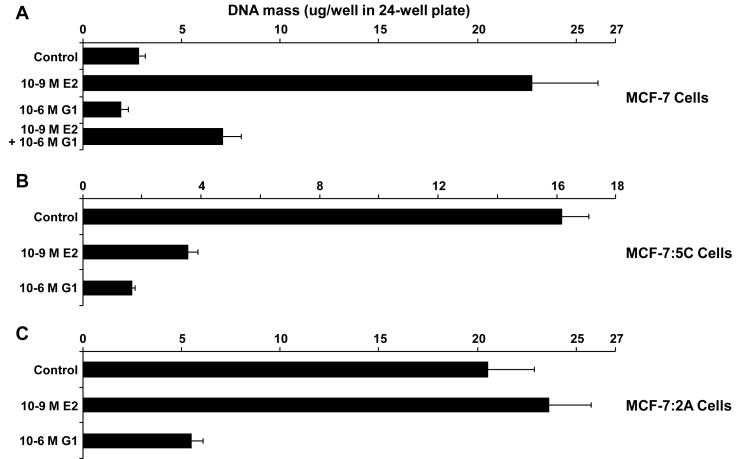
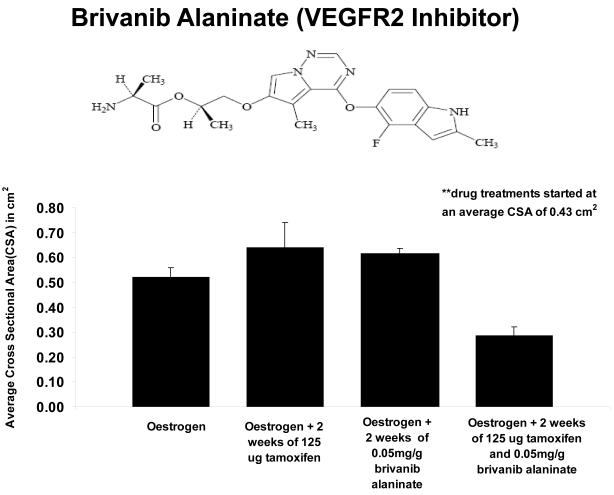
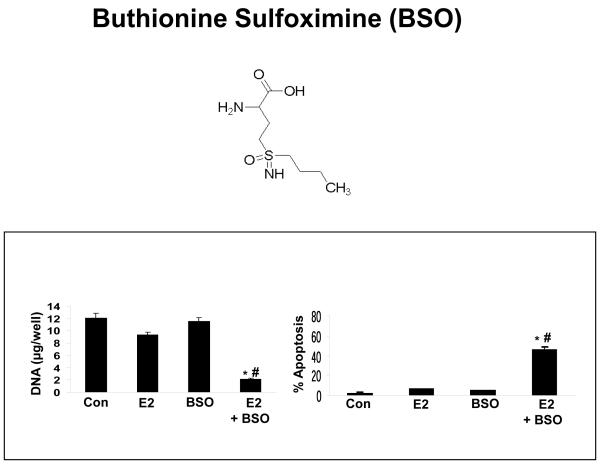
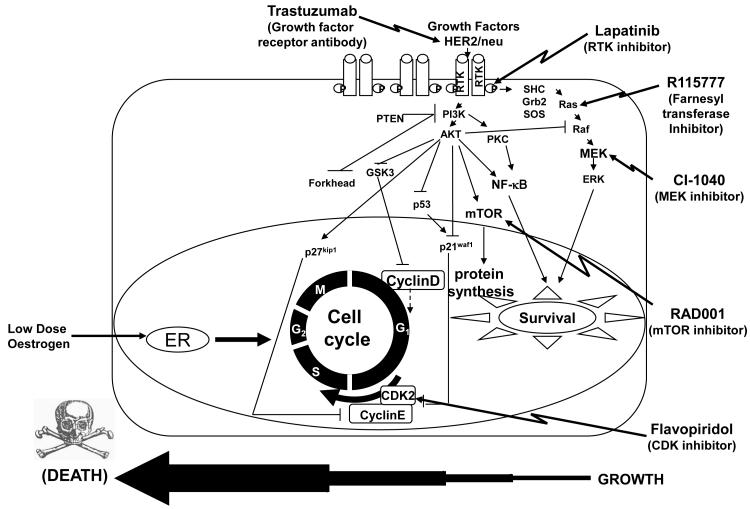
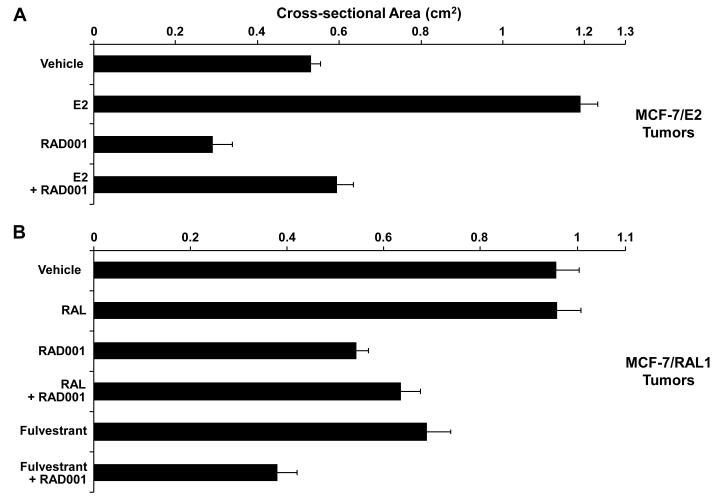
Methods and results: Three questions are posed and addressed. (1) Do we know how oestrogen works? (2) Can we improve adjuvant antihormonal therapy? (3) Can we enhance oestrogen-induced apoptosis? The new player in oestrogen action is GPR30 and there are new drugs specific for this target to trigger apoptosis. Similarly, anti-angiogenic drugs can be integrated into adjuvant antihormone therapy or to enhance oestrogen-induced apoptosis in Phase II antihormone resistant breast cancer. The goal is to reduce the development of acquired antihormone resistance or undermine the resistance of breast cancer cells to undergo apoptosis with oestrogen respectively. Finally, drugs to reduce the synthesis of glutathione, a subcellular molecule compound associated with drug resistance, can enhance oestradiol-induced apoptosis.
Conclusions: We propose an integrated approach for the rapid testing of agents to blunt survival pathways and amplify oestrogen-induced apoptosis and tumour regression in Phase II resistant metastatic breast cancer. This Pharma platform will provide rapid clinical results to predict efficacy in large scale clinical trials.
Figures

Similar articles
-
Exploiting the apoptotic actions of oestrogen to reverse antihormonal drug resistance in oestrogen receptor positive breast cancer patients.Breast. 2007 Dec;16 Suppl 2(Suppl 2):S105-13. doi: 10.1016/j.breast.2007.07.020. Epub 2007 Aug 24. Breast. 2007. PMID: 17719781 Free PMC article. Review.
-
Acquired resistance to selective estrogen receptor modulators (SERMs) in clinical practice (tamoxifen & raloxifene) by selection pressure in breast cancer cell populations.Steroids. 2014 Nov;90:44-52. doi: 10.1016/j.steroids.2014.06.002. Epub 2014 Jun 12. Steroids. 2014. PMID: 24930824 Free PMC article.
-
Where do selective estrogen receptor modulators (SERMs) and aromatase inhibitors (AIs) now fit into breast cancer treatment algorithms?J Steroid Biochem Mol Biol. 2001 Dec;79(1-5):227-37. doi: 10.1016/s0960-0760(01)00140-6. J Steroid Biochem Mol Biol. 2001. PMID: 11850229 Review.
-
Comparing the effects of 17β-oestradiol and the selective oestrogen receptor modulators, raloxifene and tamoxifen, on prepulse inhibition in female rats.Schizophr Res. 2015 Nov;168(3):634-9. doi: 10.1016/j.schres.2015.04.029. Epub 2015 Jun 5. Schizophr Res. 2015. PMID: 25979306
-
Estrogen-mediated mechanisms to control the growth and apoptosis of breast cancer cells: a translational research success story.Vitam Horm. 2013;93:1-49. doi: 10.1016/B978-0-12-416673-8.00007-1. Vitam Horm. 2013. PMID: 23810002 Review.
Cited by
-
Treatment of osteoporosis and reduction in risk of invasive breast cancer in postmenopausal women with raloxifene.Expert Opin Pharmacother. 2011 Mar;12(4):657-74. doi: 10.1517/14656566.2011.557360. Epub 2011 Feb 7. Expert Opin Pharmacother. 2011. Retraction in: Expert Opin Pharmacother. 2012 May;13(7):1081. doi: 10.1517/14656566.2012.677336. PMID: 21294695 Free PMC article. Retracted. Review.
-
Clinical benefit of sequential use of endocrine therapies for metastatic breast cancer.Int J Clin Oncol. 2015 Apr;20(2):253-61. doi: 10.1007/s10147-015-0793-8. Epub 2015 Feb 12. Int J Clin Oncol. 2015. PMID: 25673474 Review.
-
Glyceollin, a novel regulator of mTOR/p70S6 in estrogen receptor positive breast cancer.J Steroid Biochem Mol Biol. 2015 Jun;150:17-23. doi: 10.1016/j.jsbmb.2014.12.014. Epub 2015 Mar 12. J Steroid Biochem Mol Biol. 2015. PMID: 25771071 Free PMC article.
-
The estrogen receptor alpha-derived peptide ERα17p (P(295)-T(311)) exerts pro-apoptotic actions in breast cancer cells in vitro and in vivo, independently from their ERα status.Mol Oncol. 2011 Feb;5(1):36-47. doi: 10.1016/j.molonc.2010.11.001. Epub 2010 Nov 27. Mol Oncol. 2011. PMID: 21163714 Free PMC article.
-
Interaction of the Anti-Proliferative GPER Inverse Agonist ERα17p with the Breast Cancer Cell Plasma Membrane: From Biophysics to Biology.Cells. 2020 Feb 15;9(2):447. doi: 10.3390/cells9020447. Cells. 2020. PMID: 32075246 Free PMC article.
References
-
- Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nature Reviews Drug Discovery. 2003;2:205–13. - PubMed
-
- Ingle JN, Ahmann DL, Green SJ, Edmonson JH, Bisel HF, Kvols LK, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304:16–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
