

Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies
- PMID: 19915672
- PMCID: PMC2772813
- DOI: 10.1371/journal.pone.0007815
Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies
Abstract
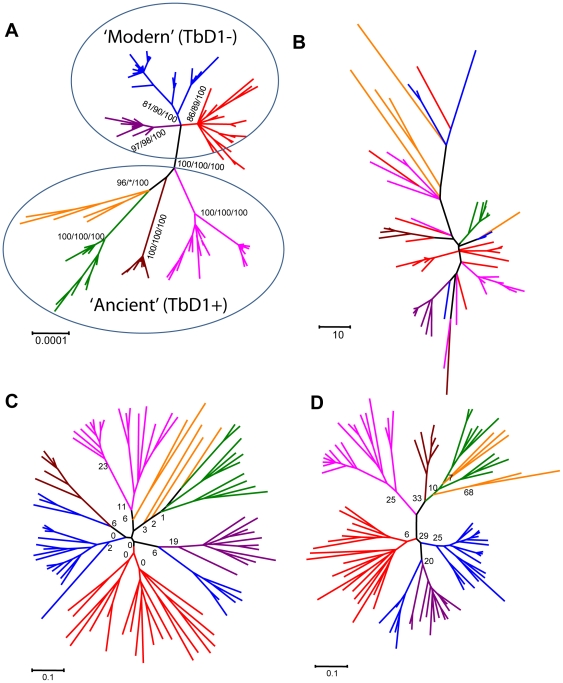
Because genetically monomorphic bacterial pathogens harbour little DNA sequence diversity, most current genotyping techniques used to study the epidemiology of these organisms are based on mobile or repetitive genetic elements. Molecular markers commonly used in these bacteria include Clustered Regulatory Short Palindromic Repeats (CRISPR) and Variable Number Tandem Repeats (VNTR). These methods are also increasingly being applied to phylogenetic and population genetic studies. Using the Mycobacterium tuberculosis complex (MTBC) as a model, we evaluated the phylogenetic accuracy of CRISPR- and VNTR-based genotyping, which in MTBC are known as spoligotyping and Mycobacterial Interspersed Repetitive Units (MIRU)-VNTR-typing, respectively. We used as a gold standard the complete DNA sequences of 89 coding genes from a global strain collection. Our results showed that phylogenetic trees derived from these multilocus sequence data were highly congruent and statistically robust, irrespective of the phylogenetic methods used. By contrast, corresponding phylogenies inferred from spoligotyping or 15-loci-MIRU-VNTR were incongruent with respect to the sequence-based trees. Although 24-loci-MIRU-VNTR performed better, it was still unable to detect all strain lineages. The DNA sequence data showed virtually no homoplasy, but the opposite was true for spoligotyping and MIRU-VNTR, which was consistent with high rates of convergent evolution and the low statistical support obtained for phylogenetic groupings defined by these markers. Our results also revealed that the discriminatory power of the standard 24 MIRU-VNTR loci varied by strain lineage. Taken together, our findings suggest strain lineages in MTBC should be defined based on phylogenetically robust markers such as single nucleotide polymorphisms or large sequence polymorphisms, and that for epidemiological purposes, MIRU-VNTR loci should be used in a lineage-dependent manner. Our findings have implications for strain typing in other genetically monomorphic bacteria.
Conflict of interest statement
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
