

HIV-1 clade B Tat, but not clade C Tat, increases X4 HIV-1 entry into resting but not activated CD4+ T cells
- PMID: 19917610
- PMCID: PMC2804326
- DOI: 10.1074/jbc.M109.049957
HIV-1 clade B Tat, but not clade C Tat, increases X4 HIV-1 entry into resting but not activated CD4+ T cells
Abstract
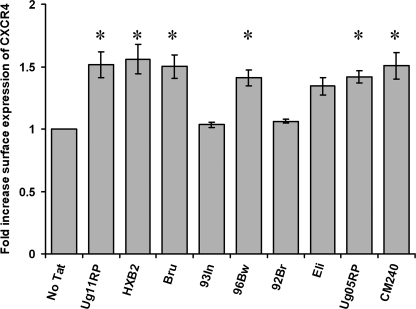
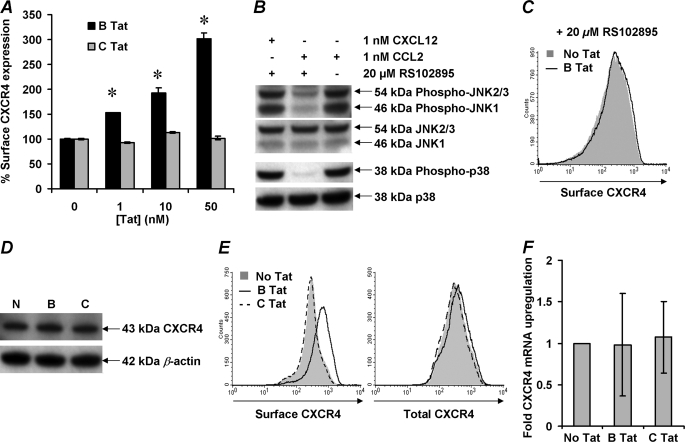
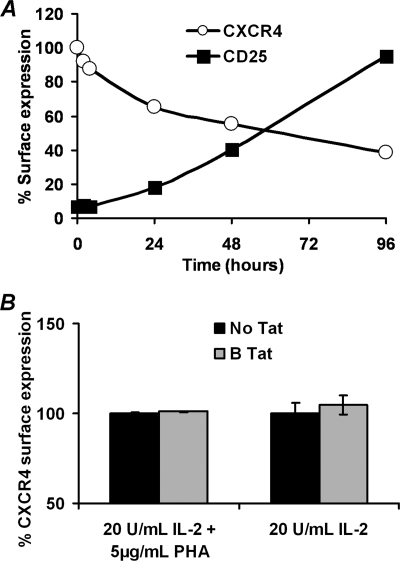
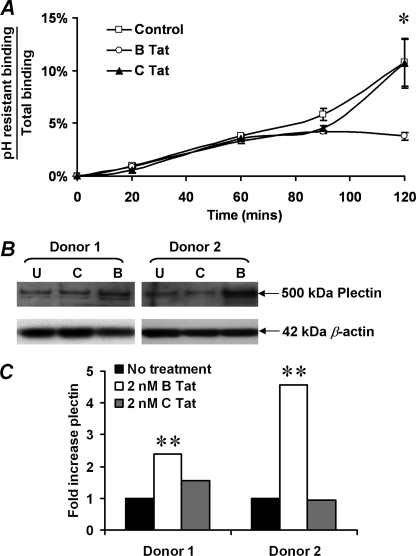
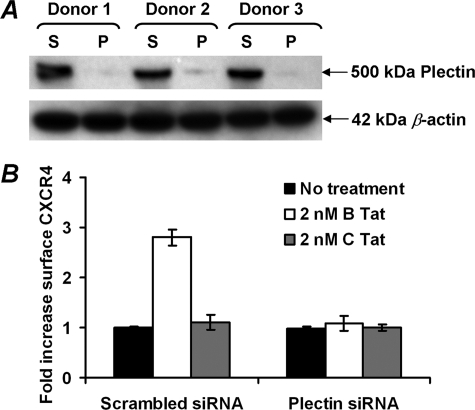
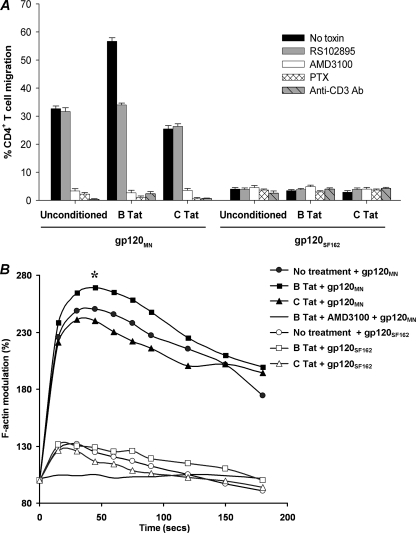
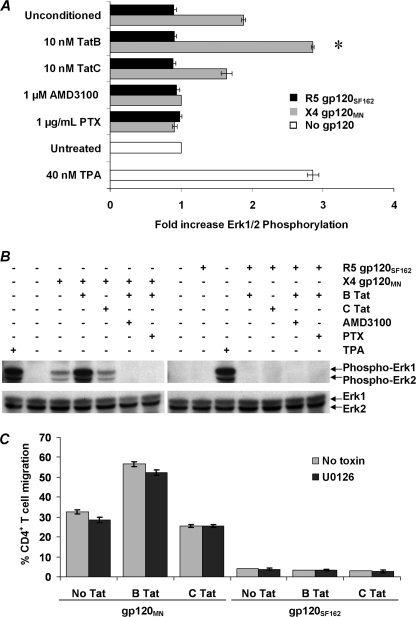
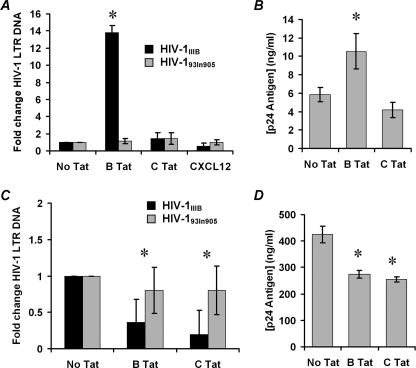
CXCR4-using human immunodeficiency virus, type 1 (HIV-1) variants emerge late in the course of infection in >40% of individuals infected with clade B HIV-1 but are described less commonly with clade C isolates. Tat is secreted by HIV-1-infected cells where it acts on both uninfected bystander cells and infected cells. In this study, we show that clade B Tat, but not clade C Tat, increases CXCR4 surface expression on resting CD4+ T cells through a CCR2b-dependent mechanism that does not involve de novo protein synthesis. The expression of plectin, a cytolinker protein that plays an important role as a scaffolding platform for proteins involved in cellular signaling including CXCR4 signaling and trafficking, was found to be significantly increased following B Tat but not C Tat treatment. Knockdown of plectin using RNA interference showed that plectin is essential for the B Tat-induced translocation of CXCR4 to the surface of resting CD4+ T cells. The increased surface CXCR4 expression following B Tat treatment led to increased function of CXCR4 including increased chemoattraction toward CXCR4-using-gp120. Moreover, increased CXCR4 surface expression rendered resting CD4+ T cells more permissive to X4 but not R5 HIV-1 infection. However, neither B Tat nor C Tat was able to up-regulate surface expression of CXCR4 on activated CD4+ T cells, and both proteins inhibited the infection of activated CD4+ T cells with X4 but not R5 HIV-1. Thus, B Tat, but not C Tat, has the capacity to render resting, but not activated, CD4+ T cells more susceptible to X4 HIV-1 infection.
Figures
Similar articles
-
Extracellular HIV-1 tat protein up-regulates the expression of surface CXC-chemokine receptor 4 in resting CD4+ T cells.J Immunol. 1999 Feb 15;162(4):2427-31. J Immunol. 1999. PMID: 9973525
-
CCL2 increases X4-tropic HIV-1 entry into resting CD4+ T cells.J Biol Chem. 2008 Nov 7;283(45):30745-53. doi: 10.1074/jbc.M804112200. Epub 2008 Sep 10. J Biol Chem. 2008. PMID: 18784079 Free PMC article.
-
Soluble HIV-1 gp120 enhances HIV-1 replication in non-dividing CD4+ T cells, mediated via cell signaling and Tat cofactor overexpression.AIDS. 2005 Jun 10;19(9):897-905. doi: 10.1097/01.aids.0000171403.07995.92. AIDS. 2005. PMID: 15905670
-
Relationships Between HIV-Mediated Chemokine Coreceptor Signaling, Cofilin Hyperactivation, Viral Tropism Switch and HIV-Mediated CD4 Depletion.Curr HIV Res. 2019;17(6):388-396. doi: 10.2174/1570162X17666191106112018. Curr HIV Res. 2019. PMID: 31702526 Review.
-
Autophagy and CD4+ T lymphocyte destruction by HIV-1.Autophagy. 2007 Jan-Feb;3(1):32-4. doi: 10.4161/auto.3275. Epub 2007 Jan 14. Autophagy. 2007. PMID: 17012832 Review.
Cited by
-
SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected macrophages.Cell Death Dis. 2020 Jul 27;11(7):590. doi: 10.1038/s41419-020-02761-x. Cell Death Dis. 2020. PMID: 32719312 Free PMC article.
-
Use of ATP analogs to inhibit HIV-1 transcription.Virology. 2012 Oct 10;432(1):219-31. doi: 10.1016/j.virol.2012.06.007. Epub 2012 Jul 6. Virology. 2012. PMID: 22771113 Free PMC article.
-
Enhanced Bone Marrow Homing of Natural Killer Cells Following mRNA Transfection With Gain-of-Function Variant CXCR4R334X.Front Immunol. 2019 Jun 5;10:1262. doi: 10.3389/fimmu.2019.01262. eCollection 2019. Front Immunol. 2019. PMID: 31231387 Free PMC article.
-
CD4+ T Cell-Mimicking Nanoparticles Broadly Neutralize HIV-1 and Suppress Viral Replication through Autophagy.mBio. 2020 Sep 15;11(5):e00903-20. doi: 10.1128/mBio.00903-20. mBio. 2020. PMID: 32934078 Free PMC article.
-
Trehalose Inhibits Human Immunodeficiency Virus Type 1 Infection in Primary Human Macrophages and CD4+ T Lymphocytes through Two Distinct Mechanisms.J Virol. 2020 Aug 17;94(17):e00237-20. doi: 10.1128/JVI.00237-20. Print 2020 Aug 17. J Virol. 2020. PMID: 32554696 Free PMC article.
References
-
- Hemelaar J., Gouws E., Ghys P. D., Osmanov S. (2006) AIDS 20, W13–W23 - PubMed
-
- Renjifo B., Gilbert P., Chaplin B., Msamanga G., Mwakagile D., Fawzi W., Essex M.the Tanzanian Vitamin and HIV Study Group (2004) AIDS 18, 1629–1636 - PubMed
-
- Bhoopat L., Rithaporn T. S., Khunamornpong S., Bhoopat T., Taylor C. R., Thorner P. S. (2006) Mod. Pathol. 19, 255–263 - PubMed
-
- Vasan A., Renjifo B., Hertzmark E., Chaplin B., Msamanga G., Essex M., Fawzi W., Hunter D. (2006) Clin. Infect. Dis. 42, 843–852 - PubMed
-
- Kaleebu P., Nankya I. L., Yirrell D. L., Shafer L. A., Kyosiimire-Lugemwa J., Lule D. B., Morgan D., Beddows S., Weber J., Whitworth J. A. (2007) J. Acquir. Immune Defic. Syndr. 45, 28–33 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
