

Estimating divergence parameters with small samples from a large number of loci
- PMID: 19917765
- PMCID: PMC2828718
- DOI: 10.1534/genetics.109.110528
Estimating divergence parameters with small samples from a large number of loci
Abstract
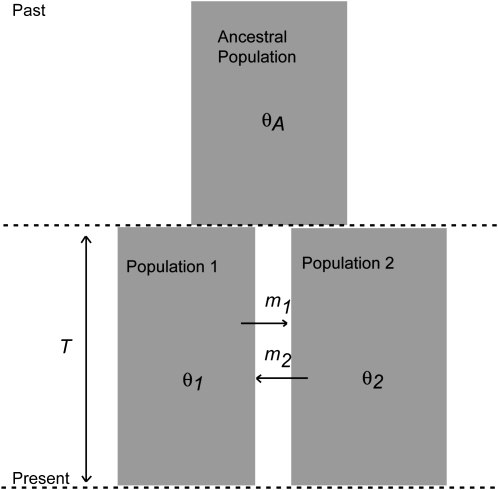
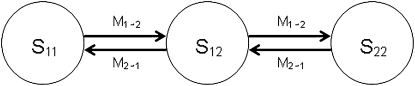
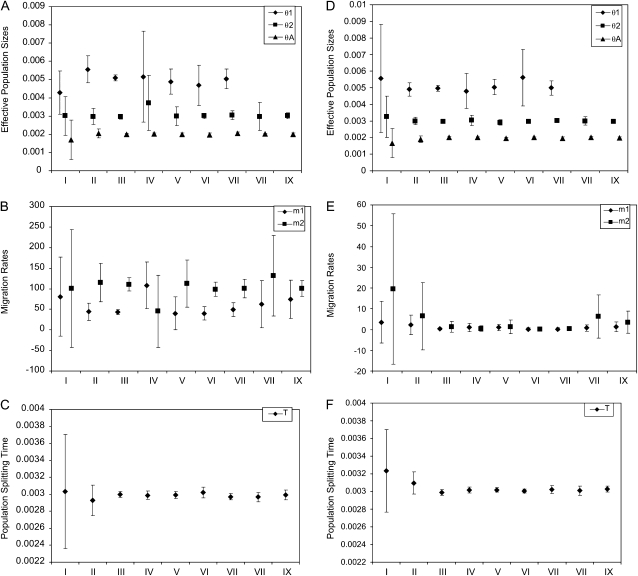
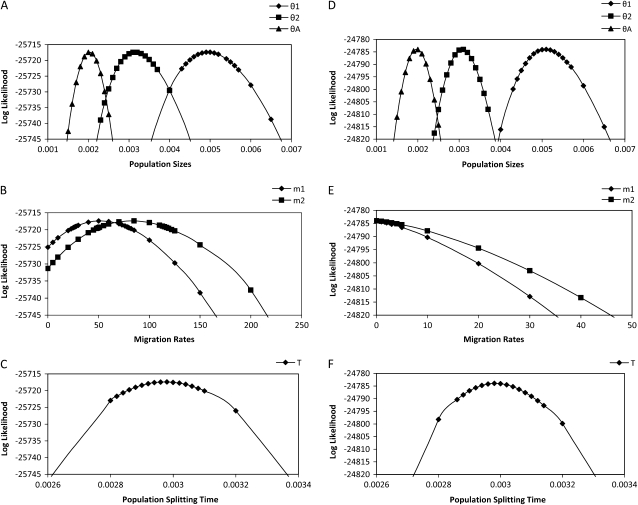
Most methods for studying divergence with gene flow rely upon data from many individuals at few loci. Such data can be useful for inferring recent population history but they are unlikely to contain sufficient information about older events. However, the growing availability of genome sequences suggests a different kind of sampling scheme, one that may be more suited to studying relatively ancient divergence. Data sets extracted from whole-genome alignments may represent very few individuals but contain a very large number of loci. To take advantage of such data we developed a new maximum-likelihood method for genomic data under the isolation-with-migration model. Unlike many coalescent-based likelihood methods, our method does not rely on Monte Carlo sampling of genealogies, but rather provides a precise calculation of the likelihood by numerical integration over all genealogies. We demonstrate that the method works well on simulated data sets. We also consider two models for accommodating mutation rate variation among loci and find that the model that treats mutation rates as random variables leads to better estimates. We applied the method to the divergence of Drosophila melanogaster and D. simulans and detected a low, but statistically significant, signal of gene flow from D. simulans to D. melanogaster.
Figures
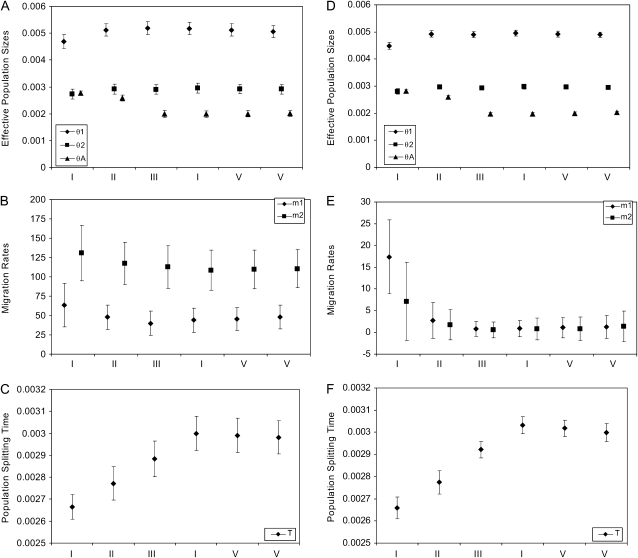
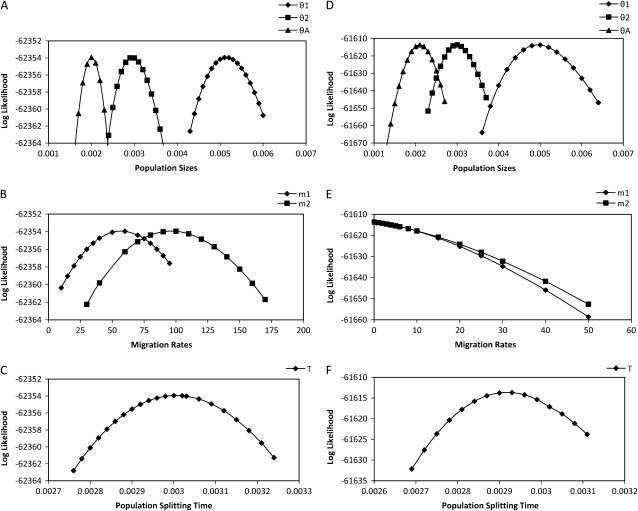
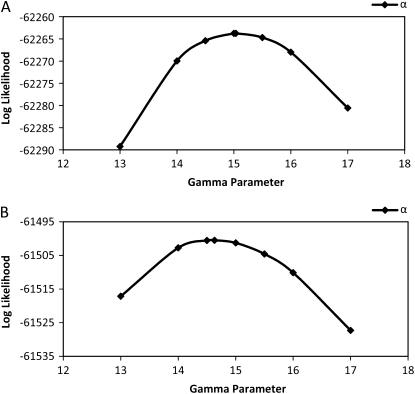
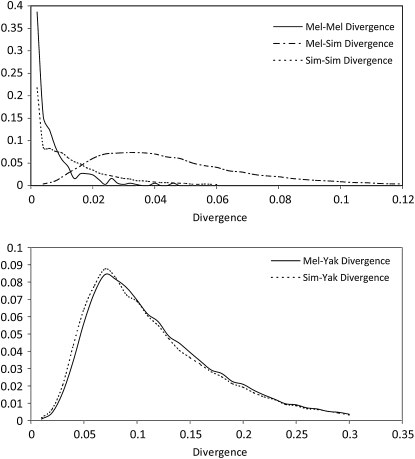
Comment in
-
On computing the coalescence time density in an isolation-with-migration model with few samples.Genetics. 2011 Apr;187(4):1241-3. doi: 10.1534/genetics.110.124164. Epub 2011 Feb 14. Genetics. 2011. PMID: 21321131 Free PMC article. No abstract available.
References
-
- Burgess, R., and Z. Yang, 2008. Estimation of hominoid ancestral population sizes under Bayesian coalescent models incorporating mutation rate variation and sequencing errors. Mol. Biol. Evol. 25 1979–1994. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
