

WNT5A mutations in patients with autosomal dominant Robinow syndrome
- PMID: 19918918
- PMCID: PMC4059519
- DOI: 10.1002/dvdy.22156
WNT5A mutations in patients with autosomal dominant Robinow syndrome
Abstract
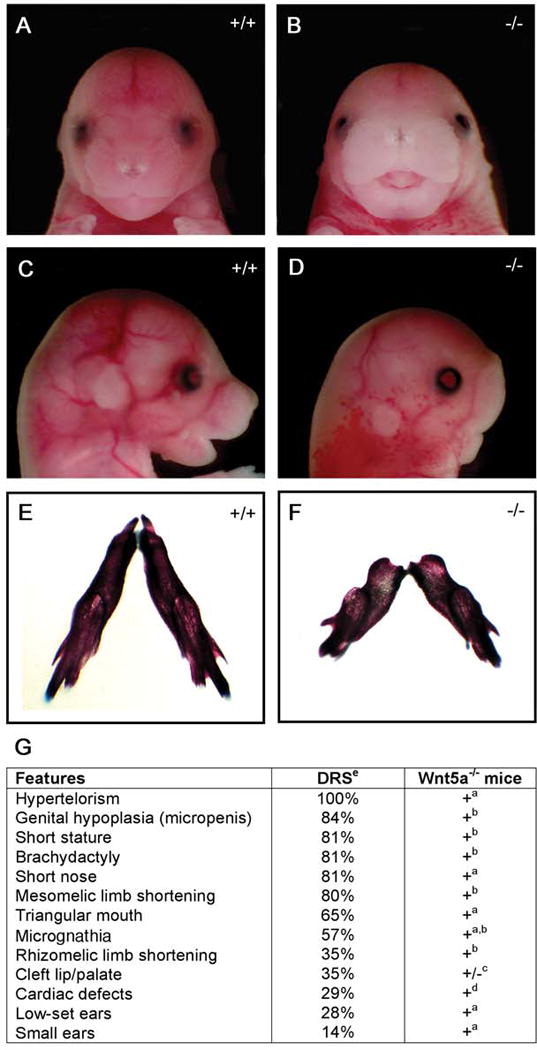
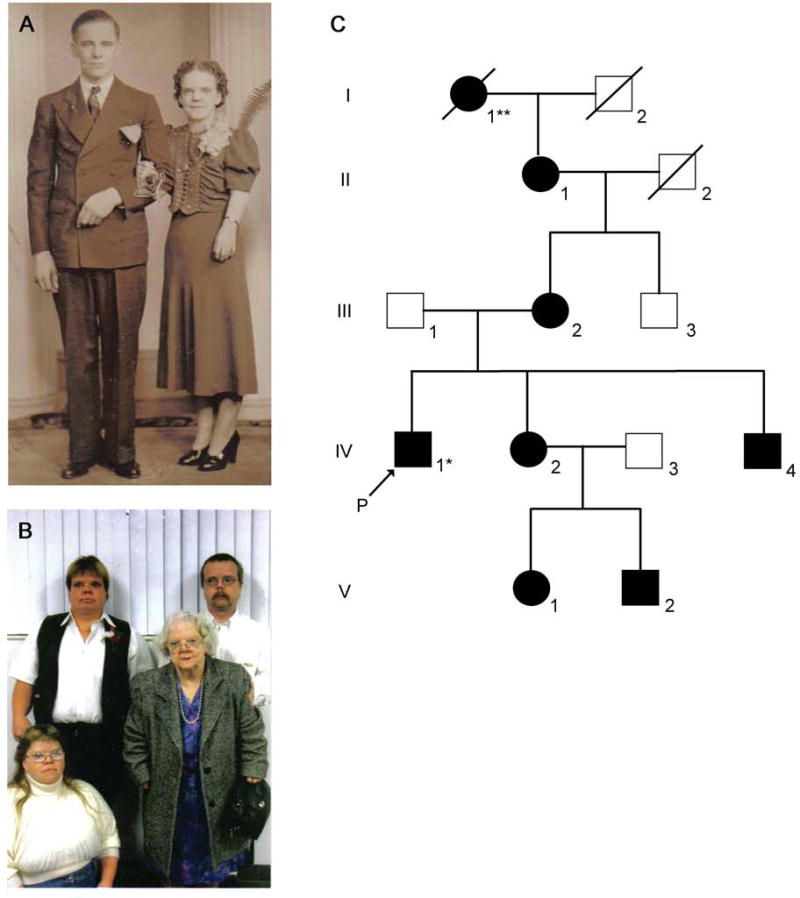
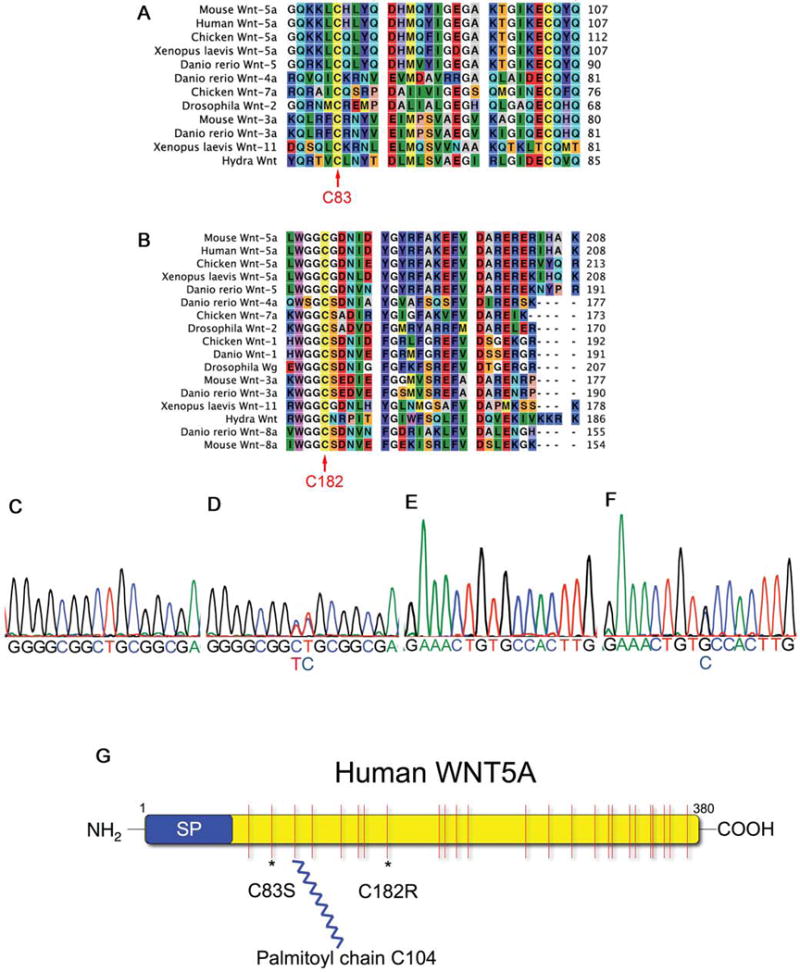
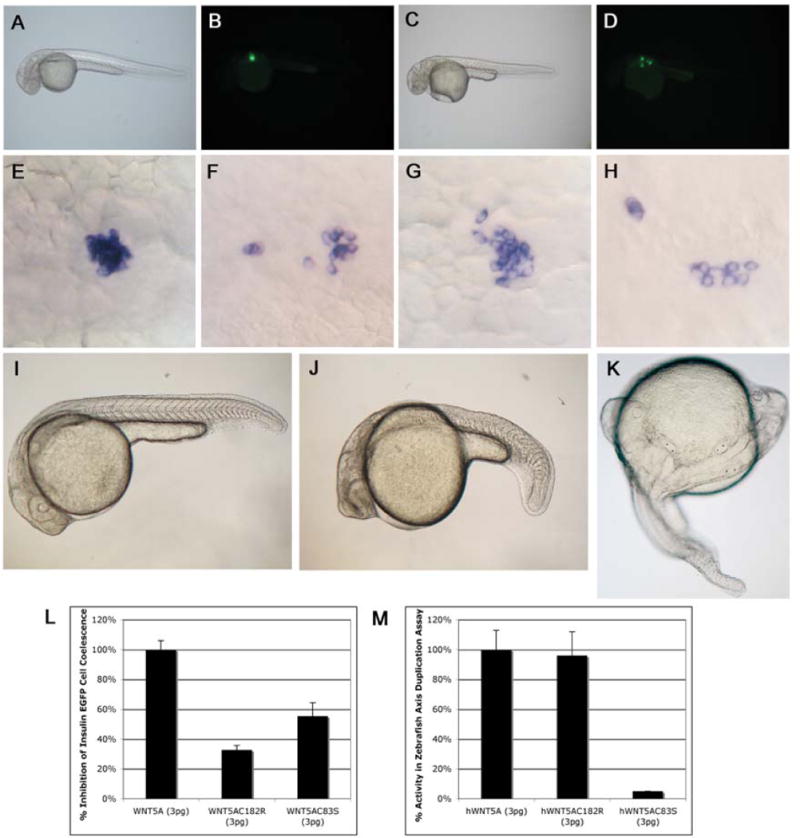
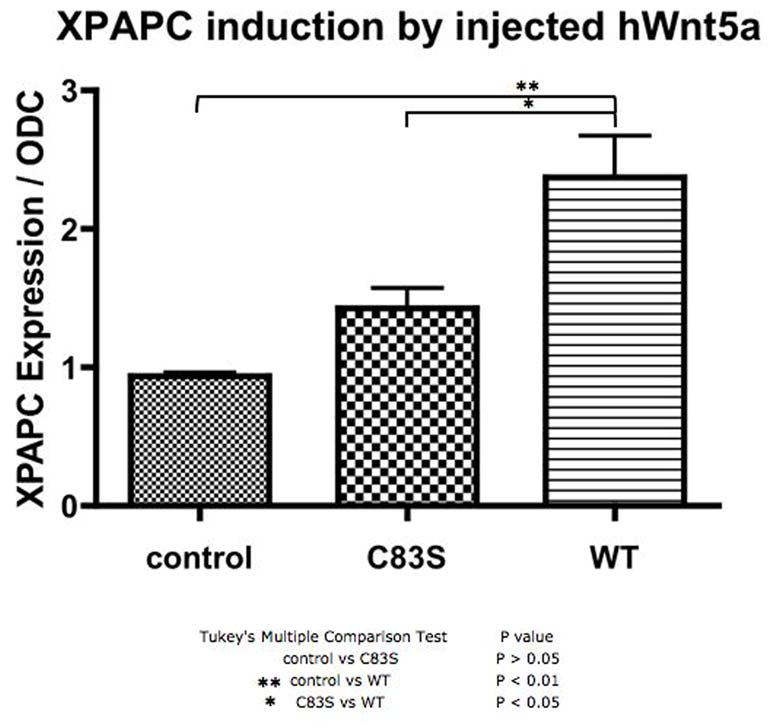
Robinow syndrome is a skeletal dysplasia with both autosomal dominant and autosomal recessive inheritance patterns. It is characterized by short stature, limb shortening, genital hypoplasia, and craniofacial abnormalities. The etiology of dominant Robinow syndrome is unknown; however, the phenotypically more severe autosomal recessive form of Robinow syndrome has been associated with mutations in the orphan tyrosine kinase receptor, ROR2, which has recently been identified as a putative WNT5A receptor. Here, we show that two different missense mutations in WNT5A, which result in amino acid substitutions of highly conserved cysteines, are associated with autosomal dominant Robinow syndrome. One mutation has been found in all living affected members of the original family described by Meinhard Robinow and another in a second unrelated patient. These missense mutations result in decreased WNT5A activity in functional assays of zebrafish and Xenopus development. This work suggests that a WNT5A/ROR2 signal transduction pathway is important in human craniofacial and skeletal development and that proper formation and growth of these structures is sensitive to variations in WNT5A function.
(c) 2009 Wiley-Liss, Inc.
Conflict of interest statement
The authors declare that they have no competing financial interests.
Figures

References
-
- Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, Tuysuz B, Murday VA, Patton MA, Wilkie AO, et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat Genet. 2000;25:419–422. - PubMed
-
- Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV, et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14:353–356. - PubMed
-
- Brugmann SA, Goodnough LH, Gregorieff A, Leucht P, Ten Berge D, Fuerer C, Clevers H, Nusse R, Helms JA. Wnt signaling mediates regional specification in the vertebrate face. Development. 2007;134:3283–3295. - PubMed
-
- DeChiara TM, Kimble RB, Poueymirou WT, Rojas J, Masiakowski P, Valenzuela DM, Yancopoulos GD. Ror2, encoding a receptor-like tyrosine kinase, is required for cartilage and growth plate development. Nat Genet. 2000;24:271–274. - PubMed
-
- Hammerschmidt M, Pelegri F, Mullins MC, Kane DA, Brand M, van Eeden FJ, Furutani-Seiki M, Granato M, Haffter P, Heisenberg CP, et al. Mutations affecting morphogenesis during gastrulation and tail formation in the zebrafish, Danio rerio. Development. 1996;123:143–151. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
