

Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth
- PMID: 19920251
- PMCID: PMC2812789
- DOI: 10.1126/scisignal.2000431
Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth
Abstract
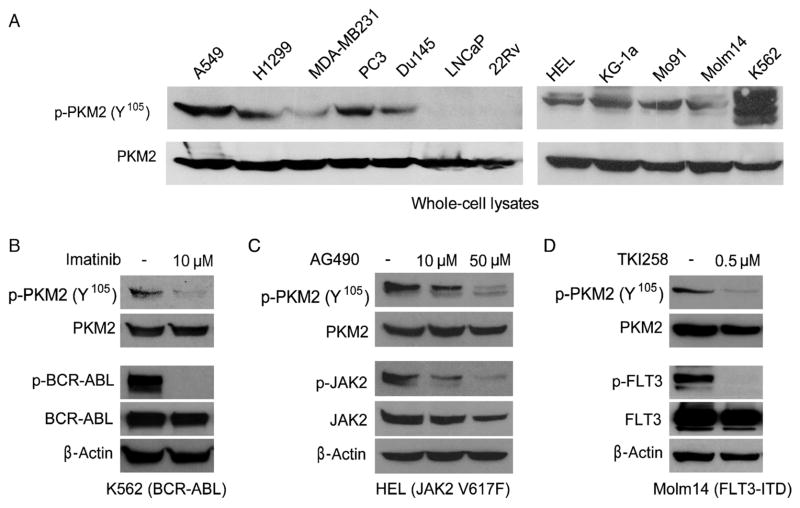
The Warburg effect describes a pro-oncogenic metabolism switch such that cancer cells take up more glucose than normal tissue and favor incomplete oxidation of glucose even in the presence of oxygen. To better understand how tyrosine kinase signaling, which is commonly increased in tumors, regulates the Warburg effect, we performed phosphoproteomic studies. We found that oncogenic forms of fibroblast growth factor receptor type 1 inhibit the pyruvate kinase M2 (PKM2) isoform by direct phosphorylation of PKM2 tyrosine residue 105 (Y(105)). This inhibits the formation of active, tetrameric PKM2 by disrupting binding of the PKM2 cofactor fructose-1,6-bisphosphate. Furthermore, we found that phosphorylation of PKM2 Y(105) is common in human cancers. The presence of a PKM2 mutant in which phenylalanine is substituted for Y(105) (Y105F) in cancer cells leads to decreased cell proliferation under hypoxic conditions, increased oxidative phosphorylation with reduced lactate production, and reduced tumor growth in xenografts in nude mice. Our findings suggest that tyrosine phosphorylation regulates PKM2 to provide a metabolic advantage to tumor cells, thereby promoting tumor growth.
Figures




References
-
- Brahimi-Horn MC, Chiche J, Pouyssegur J. Hypoxia signalling controls metabolic demand. Curr Opin Cell Biol. 2007;19:223–229. - PubMed
-
- Kroemer G, Pouyssegur J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. - PubMed
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Hathurusinghe HR, Goonetilleke KS, Siriwardena AK. Current status of tumor M2 pyruvate kinase (tumor M2-PK) as a biomarker of gastrointestinal malignancy. Ann Surg Oncol. 2007;14:2714–2720. - PubMed
-
- Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–308. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
