

alpha1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice
- PMID: 19920353
- PMCID: PMC2786799
- DOI: 10.1172/JCI39724
alpha1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice
Abstract
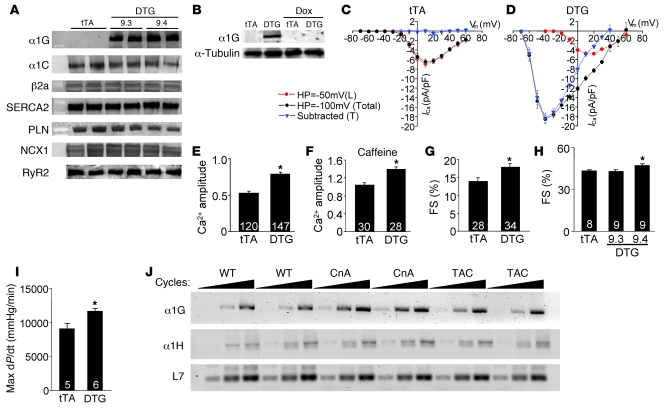
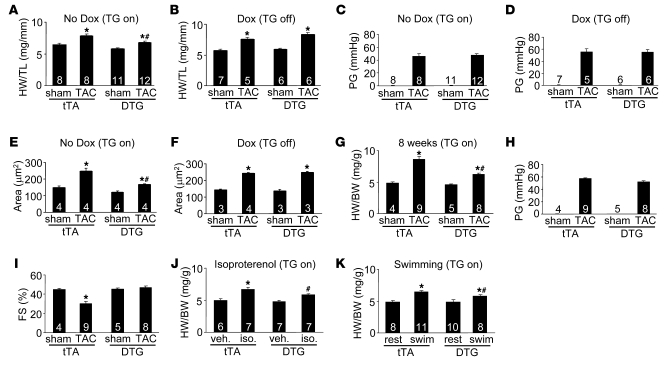
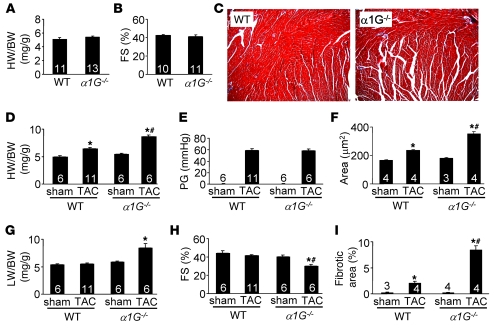
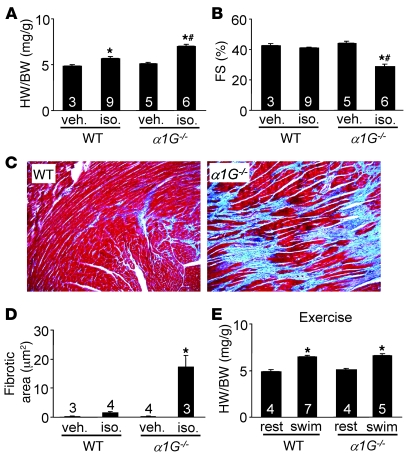
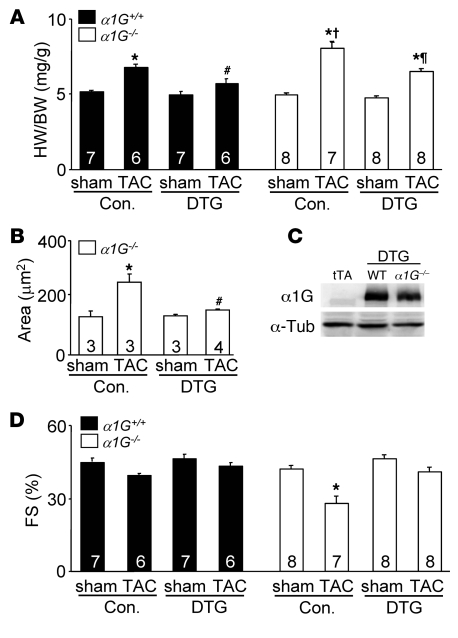
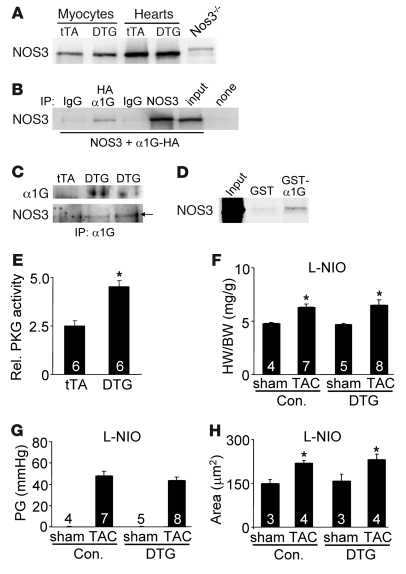
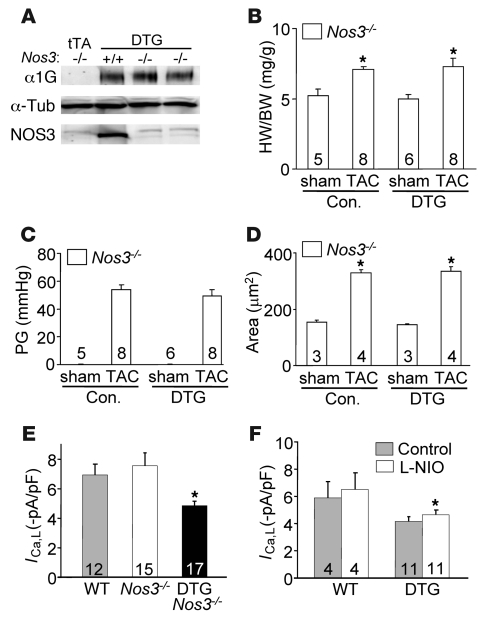
In noncontractile cells, increases in intracellular Ca2+ concentration serve as a second messenger to signal proliferation, differentiation, metabolism, motility, and cell death. Many of these Ca2+-dependent regulatory processes operate in cardiomyocytes, although it remains unclear how Ca2+ serves as a second messenger given the high Ca2+ concentrations that control contraction. T-type Ca2+ channels are reexpressed in adult ventricular myocytes during pathologic hypertrophy, although their physiologic function remains unknown. Here we generated cardiac-specific transgenic mice with inducible expression of alpha1G, which generates Cav3.1 current, to investigate whether this type of Ca2+ influx mechanism regulates the cardiac hypertrophic response. Unexpectedly, alpha1G transgenic mice showed no cardiac pathology despite large increases in Ca2+ influx, and they were even partially resistant to pressure overload-, isoproterenol-, and exercise-induced cardiac hypertrophy. Conversely, alpha1G-/- mice displayed enhanced hypertrophic responses following pressure overload or isoproterenol infusion. Enhanced hypertrophy and disease in alpha1G-/- mice was rescued with the alpha1G transgene, demonstrating a myocyte-autonomous requirement of alpha1G for protection. Mechanistically, alpha1G interacted with NOS3, which augmented cGMP-dependent protein kinase type I activity in alpha1G transgenic hearts after pressure overload. Further, the anti-hypertrophic effect of alpha1G overexpression was abrogated by a NOS3 inhibitor and by crossing the mice onto the Nos3-/- background. Thus, cardiac alpha1G reexpression and its associated pool of T-type Ca2+ antagonize cardiac hypertrophy through a NOS3-dependent signaling mechanism.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
