

MAPK phosphatase-1 facilitates the loss of oxidative myofibers associated with obesity in mice
- PMID: 19920356
- PMCID: PMC2786792
- DOI: 10.1172/JCI39054
MAPK phosphatase-1 facilitates the loss of oxidative myofibers associated with obesity in mice
Abstract
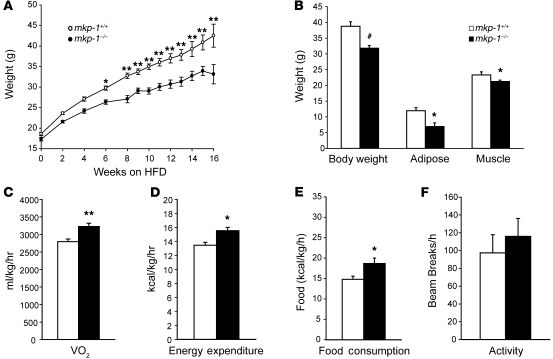
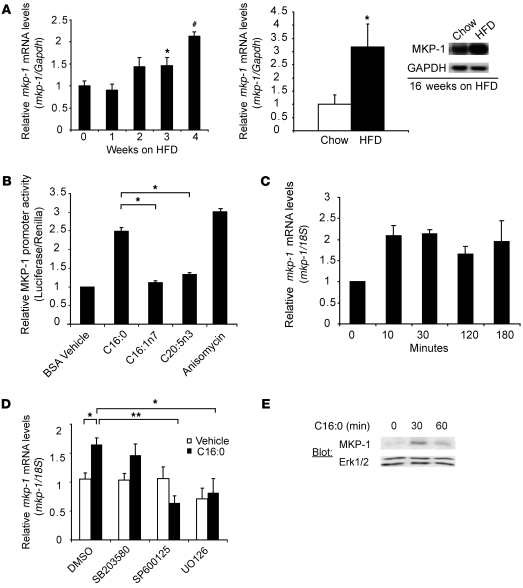
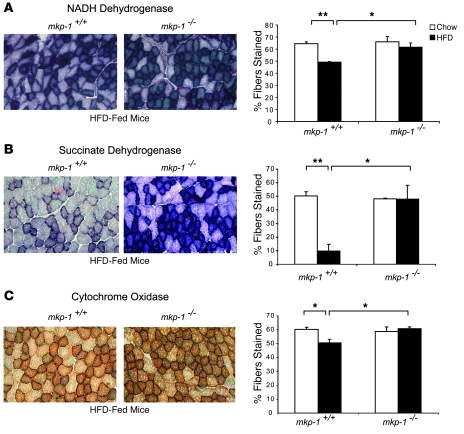
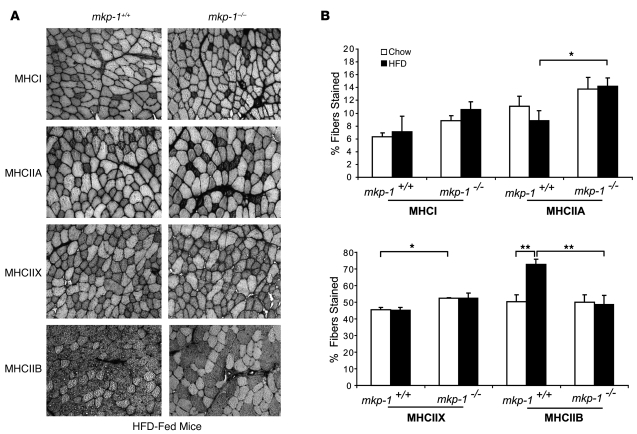
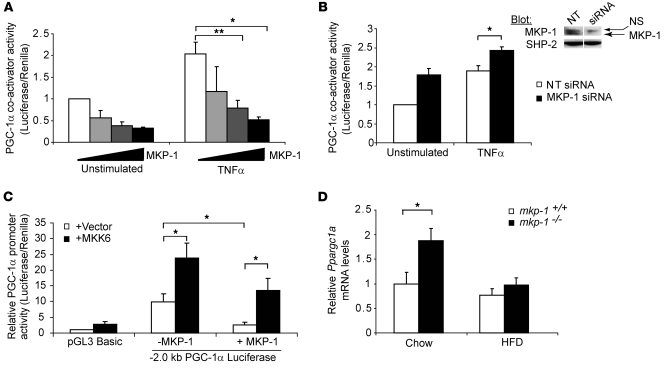
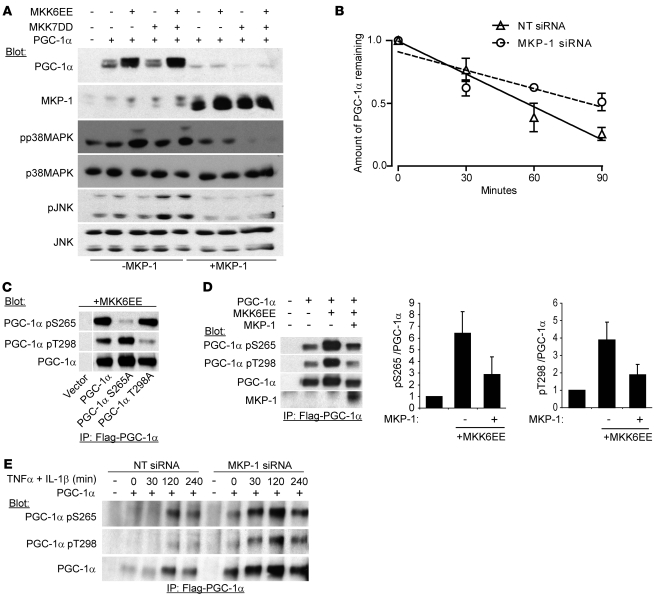
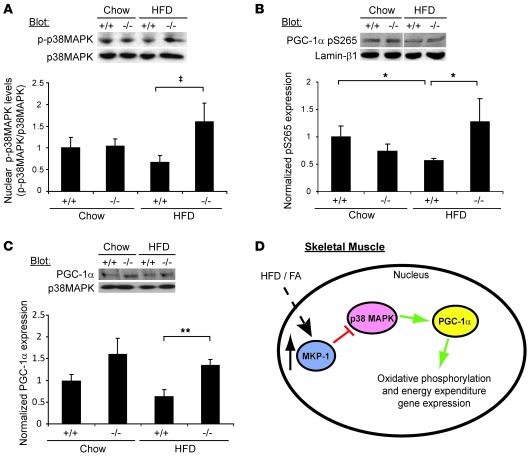
Oxidative myofibers, also known as slow-twitch myofibers, help maintain the metabolic health of mammals, and it has been proposed that decreased numbers correlate with increased risk of obesity. The transcriptional coactivator PPARgamma coactivator 1alpha (PGC-1alpha) plays a central role in maintaining levels of oxidative myofibers in skeletal muscle. Indeed, loss of PGC-1alpha expression has been linked to a reduction in the proportion of oxidative myofibers in the skeletal muscle of obese mice. MAPK phosphatase-1 (MKP-1) is encoded by mkp-1, a stress-responsive immediate-early gene that dephosphorylates MAPKs in the nucleus. Previously we showed that mice deficient in MKP-1 have enhanced energy expenditure and are resistant to diet-induced obesity. Here we show in mice that excess dietary fat induced MKP-1 overexpression in skeletal muscle, and that this resulted in reduced p38 MAPK-mediated phosphorylation of PGC-1alpha on sites that promoted its stability. Consistent with this, MKP-1-deficient mice expressed higher levels of PGC-1alpha in skeletal muscle than did wild-type mice and were refractory to the loss of oxidative myofibers when fed a high-fat diet. Collectively, these data demonstrate an essential role for MKP-1 as a regulator of the myofiber composition of skeletal muscle and suggest a potential role for MKP-1 in metabolic syndrome.
Figures
Similar articles
-
Estrogen-related receptor α regulates skeletal myocyte differentiation via modulation of the ERK MAP kinase pathway.Am J Physiol Cell Physiol. 2011 Sep;301(3):C630-45. doi: 10.1152/ajpcell.00033.2011. Epub 2011 May 11. Am J Physiol Cell Physiol. 2011. PMID: 21562305 Free PMC article.
-
Skeletal Muscle-Specific Deletion of MKP-1 Reveals a p38 MAPK/JNK/Akt Signaling Node That Regulates Obesity-Induced Insulin Resistance.Diabetes. 2018 Apr;67(4):624-635. doi: 10.2337/db17-0826. Epub 2018 Jan 9. Diabetes. 2018. PMID: 29317435 Free PMC article.
-
Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation.J Biol Chem. 2007 May 25;282(21):15439-50. doi: 10.1074/jbc.M611214200. Epub 2007 Apr 6. J Biol Chem. 2007. PMID: 17416903
-
PGC-1alpha-induced improvements in skeletal muscle metabolism and insulin sensitivity.Appl Physiol Nutr Metab. 2009 Jun;34(3):307-14. doi: 10.1139/H09-008. Appl Physiol Nutr Metab. 2009. PMID: 19448691 Review.
-
Protecting the stress response, guarding the MKP-1 mRNA.Cell Cycle. 2008 Sep 1;7(17):2640-2. doi: 10.4161/cc.7.17.6534. Epub 2008 Sep 2. Cell Cycle. 2008. PMID: 18728392 Free PMC article. Review.
Cited by
-
Skeletal muscle TET3 promotes insulin resistance through destabilisation of PGC-1α.Diabetologia. 2024 Apr;67(4):724-737. doi: 10.1007/s00125-023-06073-5. Epub 2024 Jan 13. Diabetologia. 2024. PMID: 38216792 Free PMC article.
-
Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions.Cell Mol Life Sci. 2013 Jan;70(2):223-37. doi: 10.1007/s00018-012-1041-2. Epub 2012 Jun 14. Cell Mol Life Sci. 2013. PMID: 22695679 Free PMC article. Review.
-
MAP kinase phosphatase-1 deficiency impairs skeletal muscle regeneration and exacerbates muscular dystrophy.FASEB J. 2010 Aug;24(8):2985-97. doi: 10.1096/fj.09-150045. Epub 2010 Apr 6. FASEB J. 2010. PMID: 20371627 Free PMC article.
-
Screening of Genes Related to Growth, Development and Meat Quality of Sahan Crossbred F1 Sheep Based on RNA-Seq Technology.Front Vet Sci. 2022 Apr 7;9:831519. doi: 10.3389/fvets.2022.831519. eCollection 2022. Front Vet Sci. 2022. PMID: 35464379 Free PMC article.
-
Estrogen-related receptor α regulates skeletal myocyte differentiation via modulation of the ERK MAP kinase pathway.Am J Physiol Cell Physiol. 2011 Sep;301(3):C630-45. doi: 10.1152/ajpcell.00033.2011. Epub 2011 May 11. Am J Physiol Cell Physiol. 2011. PMID: 21562305 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- U24 DK076169/DK/NIDDK NIH HHS/United States
- T32 DK007356/DK/NIDDK NIH HHS/United States
- P30 DK45735/DK/NIDDK NIH HHS/United States
- U24 DK059635/DK/NIDDK NIH HHS/United States
- AR46524/AR/NIAMS NIH HHS/United States
- U24 DK-76169/DK/NIDDK NIH HHS/United States
- R01 AR046524/AR/NIAMS NIH HHS/United States
- DK75776/DK/NIDDK NIH HHS/United States
- T32 DK07356/DK/NIDDK NIH HHS/United States
- R01 DK040936/DK/NIDDK NIH HHS/United States
- R01 DK075776/DK/NIDDK NIH HHS/United States
- P30 DK045735/DK/NIDDK NIH HHS/United States
- DK40936/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
