

TGF-β1 pathway as a new target for neuroprotection in Alzheimer's disease
- PMID: 19925479
- PMCID: PMC6493850
- DOI: 10.1111/j.1755-5949.2009.00115.x
TGF-β1 pathway as a new target for neuroprotection in Alzheimer's disease
Abstract
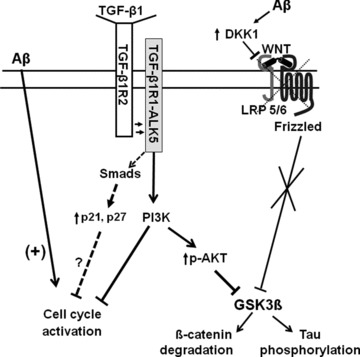
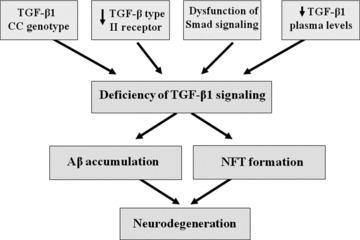
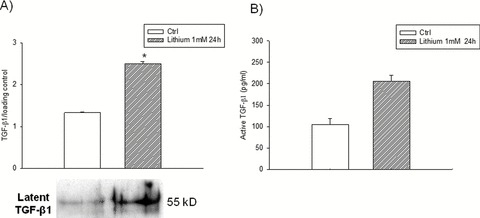
Alzheimer's disease (AD) is a neurodegenerative disorder that affects more than 37 million people worldwide. Current drugs for AD are only symptomatic, but do not interfere with the underlying pathogenic mechanisms of the disease. AD is characterized by the presence of ß-amyloid (Aβ) plaques, neurofibrillary tangles, and neuronal loss. The identification of the molecular determinants underlying AD pathogenesis is a fundamental step to design new disease-modifying drugs. Recently, a specific impairment of transforming-growth-factor-β1 (TGF-β1) signaling pathway has been demonstrated in AD brain. The deficiency of TGF-β1 signaling has been shown to increase both Aβ accumulation and Aβ-induced neurodegeneration in AD models. The loss of function of TGF-ß1 pathway seems also to contribute to tau pathology and neurofibrillary tangle formation. Growing evidence suggests a neuroprotective role for TGF-β1 against Aβ toxicity both in vitro and in vivo models of AD. Different drugs, such as lithium or group II mGlu receptor agonists are able to increase TGF-β1 levels in the central nervous system (CNS), and might be considered as new neuroprotective tools against Aβ-induced neurodegeneration. In the present review, we examine the evidence for a neuroprotective role of TGF-β1 in AD, and discuss the TGF-β1 signaling pathway as a new pharmacological target for the treatment of AD.
© 2009 Blackwell Publishing Ltd.
Conflict of interest statement
All authors declare that no potential conflict of interest exists, including all relevant financial interests in any company or institution that might benefit from the publication.
Figures
References
-
- Ballard C, Day S, Sharp S, Wing G, Sorensen S. Neuropsychiatric symptoms in dementia: Importance and treatment considerations. Int Rev Psychiatry 2008;20:396–404. - PubMed
-
- Mount C, Downton C. Alzheimer's disease: Progress or profit? Nature Med 2006;12:780–784. - PubMed
-
- Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J. Therapeutic approaches to Alzheimer's disease. Brain 2006;129:2840–2855. - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002;297:353–356. - PubMed
-
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 2005;280:17294–17300. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
