

The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis
- PMID: 19926015
- PMCID: PMC2880864
- DOI: 10.1016/j.jacc.2009.08.022
The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis
Abstract
Objectives: This study was undertaken to determine the spectrum and prevalence of mutations in the RYR2-encoded cardiac ryanodine receptor in cases with exertional syncope and normal corrected QT interval (QTc).
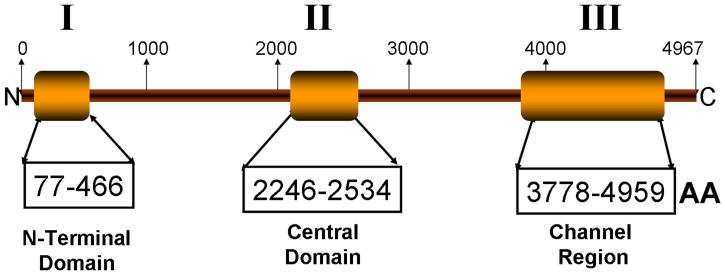
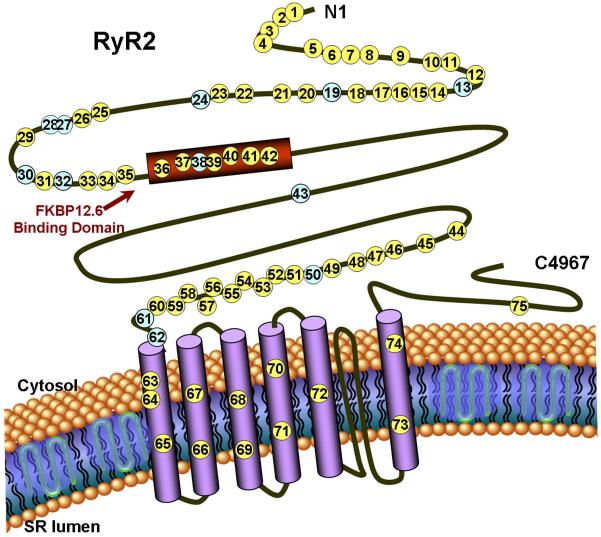
Background: Mutations in RYR2 cause type 1 catecholaminergic polymorphic ventricular tachycardia (CPVT1), a cardiac channelopathy with increased propensity for lethal ventricular dysrhythmias. Most RYR2 mutational analyses target 3 canonical domains encoded by <40% of the translated exons. The extent of CPVT1-associated mutations localizing outside of these domains remains unknown as RYR2 has not been examined comprehensively in most patient cohorts.
Methods: Mutational analysis of all RYR2 exons was performed using polymerase chain reaction, high-performance liquid chromatography, and deoxyribonucleic acid sequencing on 155 unrelated patients (49% females, 96% Caucasian, age at diagnosis 20 +/- 15 years, mean QTc 428 +/- 29 ms), with either clinical diagnosis of CPVT (n = 110) or an initial diagnosis of exercise-induced long QT syndrome but with QTc <480 ms and a subsequent negative long QT syndrome genetic test (n = 45).
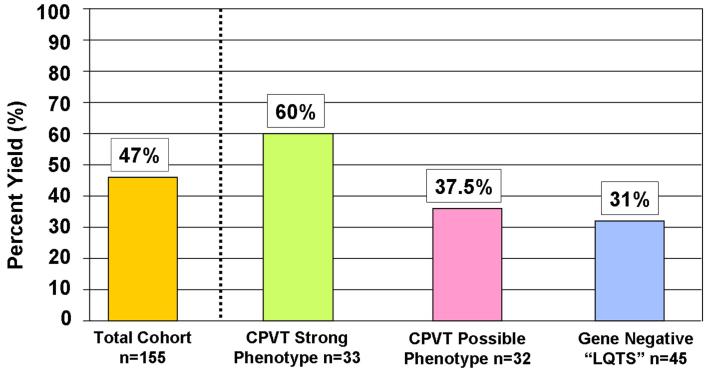
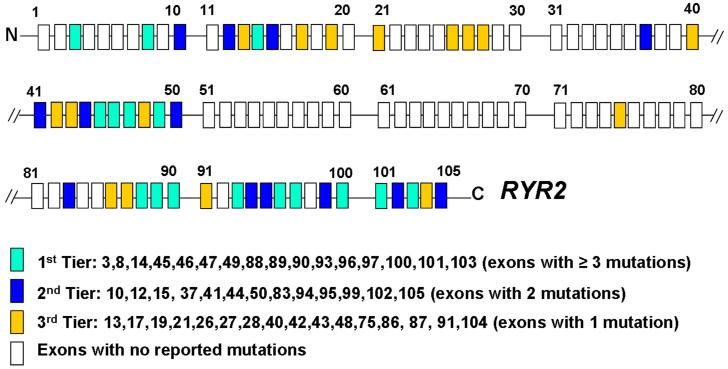
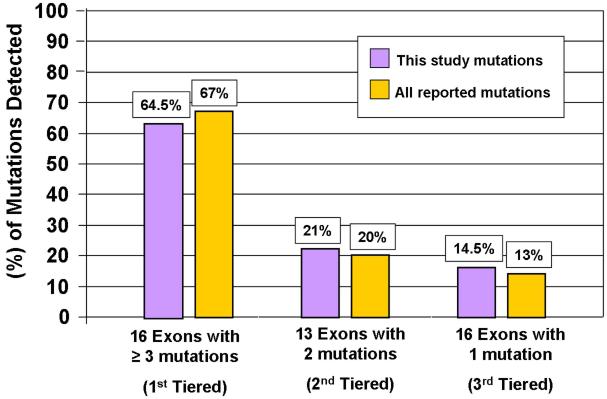
Results: Sixty-three (34 novel) possible CPVT1-associated mutations, absent in 400 reference alleles, were detected in 73 unrelated patients (47%). Thirteen new mutation-containing exons were identified. Two-thirds of the CPVT1-positive patients had mutations that localized to 1 of 16 exons.
Conclusions: Possible CPVT1 mutations in RYR2 were identified in nearly one-half of this cohort; 45 of the 105 translated exons are now known to host possible mutations. Considering that approximately 65% of CPVT1-positive cases would be discovered by selective analysis of 16 exons, a tiered targeting strategy for CPVT genetic testing should be considered.
Figures
References
-
- Leenhardt A, Lucet V, Denjoy I, et al. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–1519. - PubMed
-
- Swan H, Piippo K, Viitasalo M, et al. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035–2042. - PubMed
-
- Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. - PubMed
-
- Postma AV, Denjoy I, Hoorntje TM, et al. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–26. - PubMed
-
- Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
