

Homozygous SLC2A9 mutations cause severe renal hypouricemia
- PMID: 19926891
- PMCID: PMC2799278
- DOI: 10.1681/ASN.2009040406
Homozygous SLC2A9 mutations cause severe renal hypouricemia
Abstract
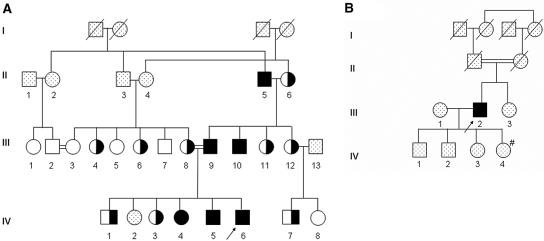
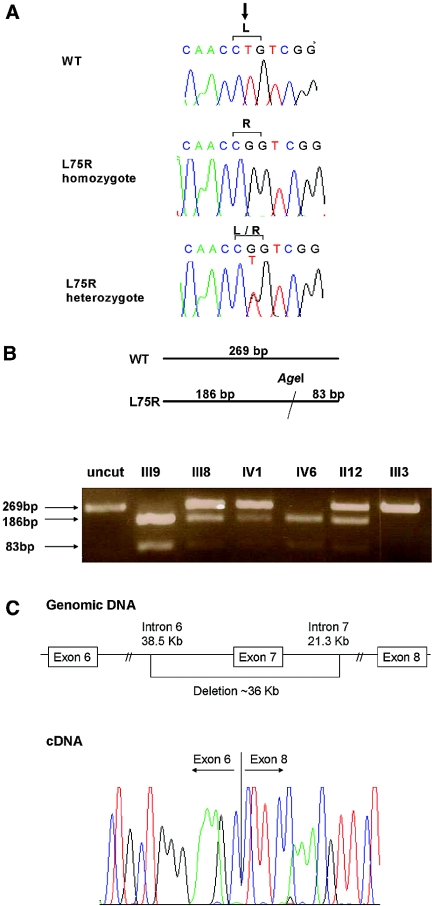
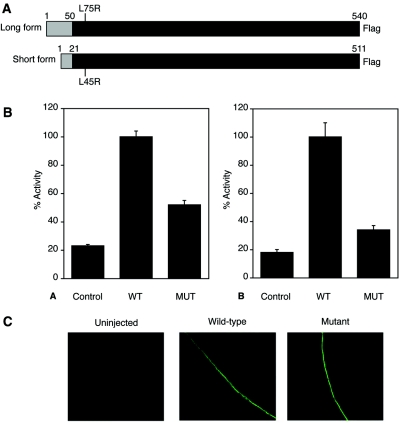
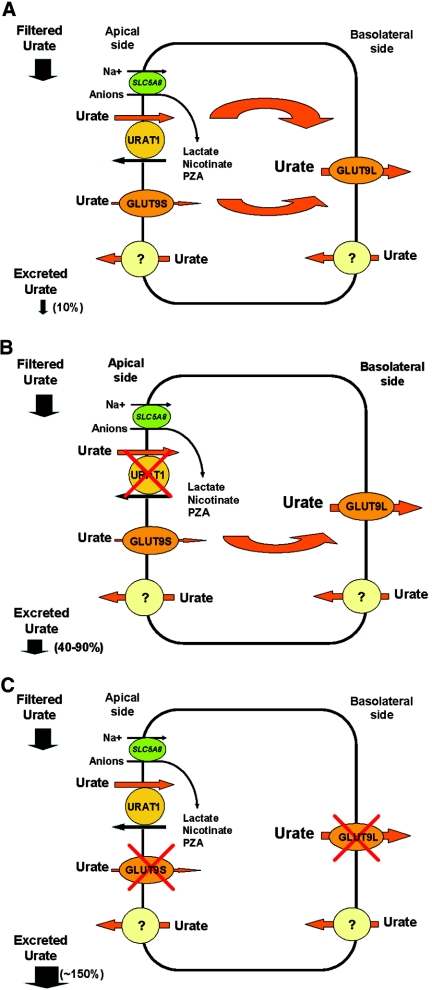
Hereditary hypouricemia may result from mutations in the renal tubular uric acid transporter URAT1. Whether mutation of other uric acid transporters produces a similar phenotype is unknown. We studied two families who had severe hereditary hypouricemia and did not have a URAT1 defect. We performed a genome-wide homozygosity screen and linkage analysis and identified the candidate gene SLC2A9, which encodes the glucose transporter 9 (GLUT9). Both families had homozygous SLC2A9 mutations: A missense mutation (L75R) in six affected members of one family and a 36-kb deletion, resulting in a truncated protein, in the other. In vitro, the L75R mutation dramatically impaired transport of uric acid. The mean concentration of serum uric acid of seven homozygous individuals was 0.17 +/- 0.2 mg/dl, and all had a fractional excretion of uric acid >150%. Three individuals had nephrolithiasis, and three had a history of exercise-induced acute renal failure. In conclusion, homozygous loss-of-function mutations of GLUT9 cause a total defect of uric acid absorption, leading to severe renal hypouricemia complicated by nephrolithiasis and exercise-induced acute renal failure. In addition to clarifying renal handling of uric acid, our findings may provide a better understanding of the pathophysiology of acute renal failure, nephrolithiasis, hyperuricemia, and gout.
Figures
References
-
- Sperling O: Hereditary renal hypouricemia. Mol Genet Metab 89: 14–18, 2006 - PubMed
-
- Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H: Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 417: 447–452, 2002 - PubMed
-
- Ichida K, Hosoyamada M, Hisatome I, Enomoto A, Hikita M, Endou H, Hosoya T: Clinical and molecular analysis of patients with renal hypouricemia in Japan: Influence of URAT1 gene on urinary urate excretion. J Am Soc Nephrol 15: 164–173, 2004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
