

Antiviral activity of the interferon-induced cellular protein BST-2/tetherin
- PMID: 19929170
- PMCID: PMC2858902
- DOI: 10.1089/aid.2009.0253
Antiviral activity of the interferon-induced cellular protein BST-2/tetherin
Abstract
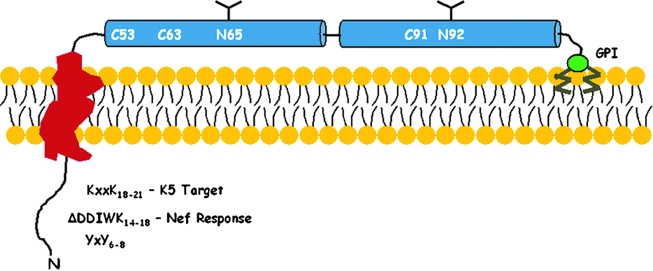
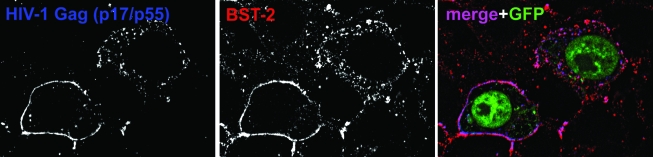
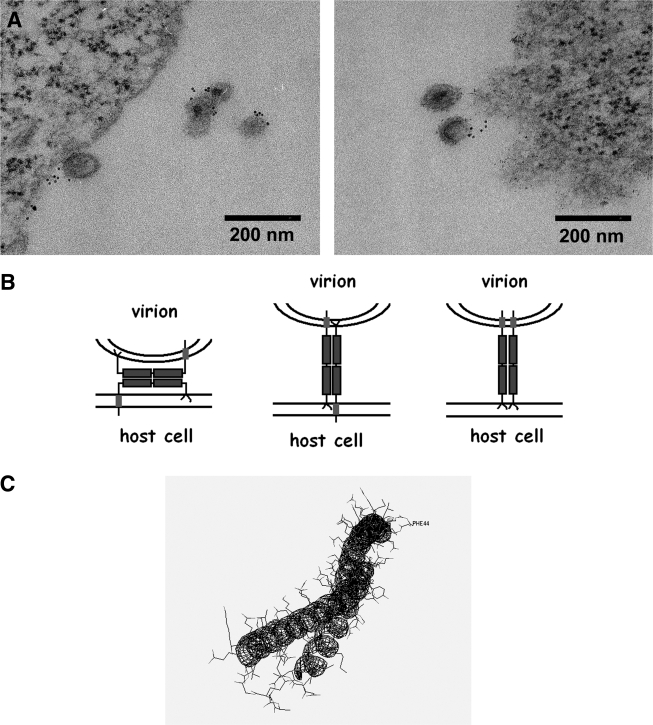
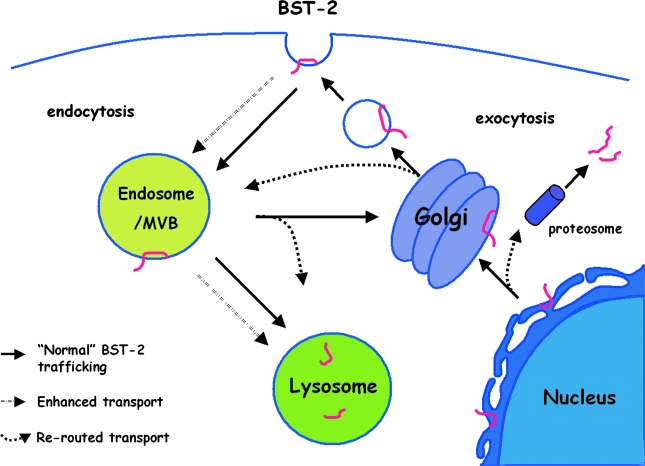
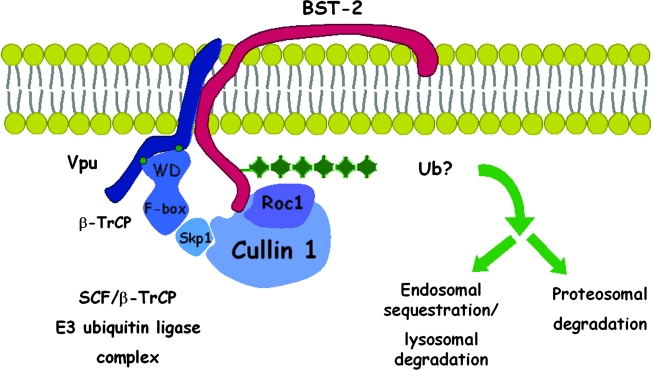
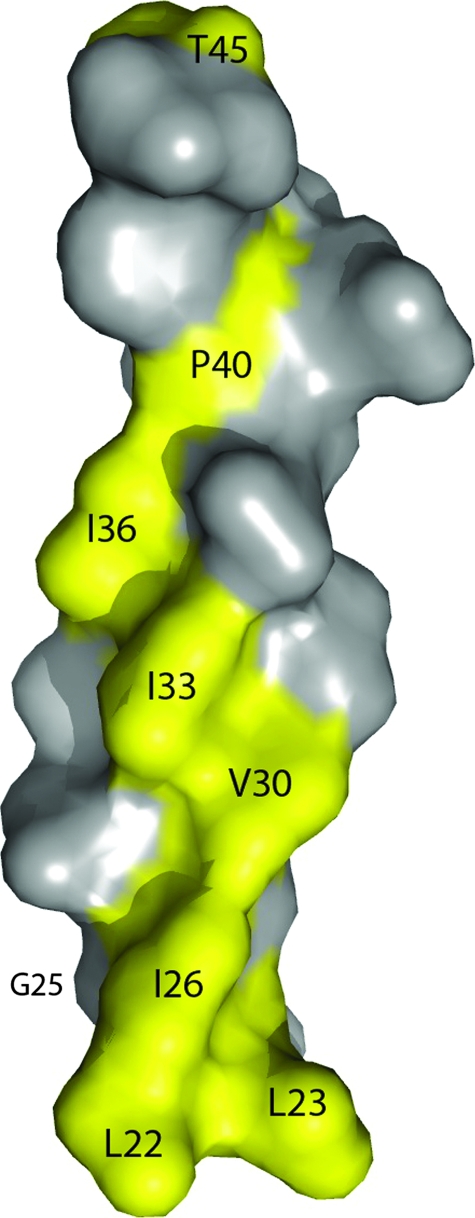
Pathogenic microorganisms encode proteins that antagonize specific aspects of innate or adaptive immunity. Just as the study of the HIV-1 accessory protein Vif led to the identification of cellular cytidine deaminases as host defense proteins, the study of HIV-1 Vpu recently led to the discovery of the interferon-induced transmembrane protein BST-2 (CD317; tetherin) as a novel component of the innate defense against enveloped viruses. BST-2 is an unusually structured protein that restricts the release of fully formed progeny virions from infected cells, presumably by a direct retention mechanism that is independent of any viral protein target. Its spectrum of activity includes at least four virus families: retroviruses, filoviruses, arenaviruses, and herpesviruses. Viral antagonists of BST-2 include HIV-1 Vpu, HIV-2 and SIV Env, SIV Nef, the Ebola envelope glycoprotein, and the K5 protein of KSHV. The mechanisms of antagonism are diverse and currently include viral cooption of cellular endosomal trafficking and protein degradation pathways, including those mediated by ubiquitination. Orthologs of human BST-2 are present in mammals. Primate BST-2 proteins are differentially sensitive to antagonism by lentiviral Vpu and Nef proteins, suggesting that BST-2 has subjected lentiviruses to evolutionary pressure and presents barriers to cross-species transmission. BST-2 functions not only as an effector of the interferon-induced antiviral response but also as a negative feedback regulator of interferon production by plasmacytoid dendritic cells. Future work will focus on the role and regulation of BST-2 during the innate response to viral infection, on the mechanisms of restriction and of antagonism by viral gene products, and on the role of BST-2 in primate lentiviral evolution. The augmentation of BST-2 activity and the inhibition of virally encoded antagonists, in particular Vpu, represent new approaches to the prevention and treatment of HIV-1 infection.
Figures
Similar articles
-
Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2.PLoS Pathog. 2009 May;5(5):e1000429. doi: 10.1371/journal.ppat.1000429. Epub 2009 May 15. PLoS Pathog. 2009. PMID: 19436700 Free PMC article.
-
Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles.PLoS Pathog. 2010 Mar 5;6(3):e1000701. doi: 10.1371/journal.ppat.1000701. PLoS Pathog. 2010. PMID: 20221443 Free PMC article.
-
Vpu of a Simian Immunodeficiency Virus Isolated from Greater Spot-Nosed Monkey Antagonizes Human BST-2 via Two AxxxxxxxW Motifs.J Virol. 2020 Jan 6;94(2):e01669-19. doi: 10.1128/JVI.01669-19. Print 2020 Jan 6. J Virol. 2020. PMID: 31666374 Free PMC article.
-
Emerging role of the host restriction factor tetherin in viral immune sensing.J Mol Biol. 2013 Dec 13;425(24):4956-64. doi: 10.1016/j.jmb.2013.09.029. Epub 2013 Sep 26. J Mol Biol. 2013. PMID: 24075872 Review.
-
BST-2/tetherin: a new component of the innate immune response to enveloped viruses.Trends Microbiol. 2010 Sep;18(9):388-96. doi: 10.1016/j.tim.2010.06.010. Epub 2010 Aug 3. Trends Microbiol. 2010. PMID: 20688520 Free PMC article. Review.
Cited by
-
Regulation of Interferon-Stimulated Gene BST2 by a lncRNA Transcribed from a Shared Bidirectional Promoter.Front Immunol. 2015 Jan 30;5:676. doi: 10.3389/fimmu.2014.00676. eCollection 2014. Front Immunol. 2015. PMID: 25688240 Free PMC article.
-
Ebola virus glycoprotein counteracts BST-2/Tetherin restriction in a sequence-independent manner that does not require tetherin surface removal.J Virol. 2010 Jul;84(14):7243-55. doi: 10.1128/JVI.02636-09. Epub 2010 May 5. J Virol. 2010. PMID: 20444895 Free PMC article.
-
Ion channels as antivirus targets.Virol Sin. 2010 Aug;25(4):267-80. doi: 10.1007/s12250-010-3136-y. Epub 2010 Jul 28. Virol Sin. 2010. PMID: 20960300 Free PMC article. Review.
-
Evidence for the innate immune response as a correlate of protection in human immunodeficiency virus (HIV)-1 highly exposed seronegative subjects (HESN).Clin Exp Immunol. 2011 May;164(2):158-69. doi: 10.1111/j.1365-2249.2011.04379.x. Epub 2011 Mar 17. Clin Exp Immunol. 2011. PMID: 21413945 Free PMC article. Review.
-
Host factors facilitating SARS-CoV-2 virus infection and replication in the lungs.Cell Mol Life Sci. 2021 Aug;78(16):5953-5976. doi: 10.1007/s00018-021-03889-5. Epub 2021 Jul 5. Cell Mol Life Sci. 2021. PMID: 34223911 Free PMC article. Review.
References
-
- Strebel K. Klimkait T. Martin MA. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science. 1988;241:1221–1223. - PubMed
-
- Cohen EA. Terwilliger EF. Sodroski JG. Haseltine WA. Identification of a protein encoded by the vpu gene of HIV-1. Nature. 1988;334:532–534. - PubMed
-
- Sakai H. Tokunaga K. Kawamura M. Adachi A. Function of human immunodeficiency virus type 1 Vpu protein in various cell types. J Gen Virol. 1995;76(Pt 11):2717–2722. - PubMed
-
- Yasuda Y. Miyake S. Kato S, et al. Interferon-alpha treatment leads to accumulation of virus particles on the surface of cells persistently infected with the human immunodeficiency virus type 1. J Acquir Immune Defic Syndr. 1990;3(11):1046–1051. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
