

Global changes in processing of mRNA 3' untranslated regions characterize clinically distinct cancer subtypes
- PMID: 19934316
- PMCID: PMC2794997
- DOI: 10.1158/0008-5472.CAN-09-2236
Global changes in processing of mRNA 3' untranslated regions characterize clinically distinct cancer subtypes
Abstract
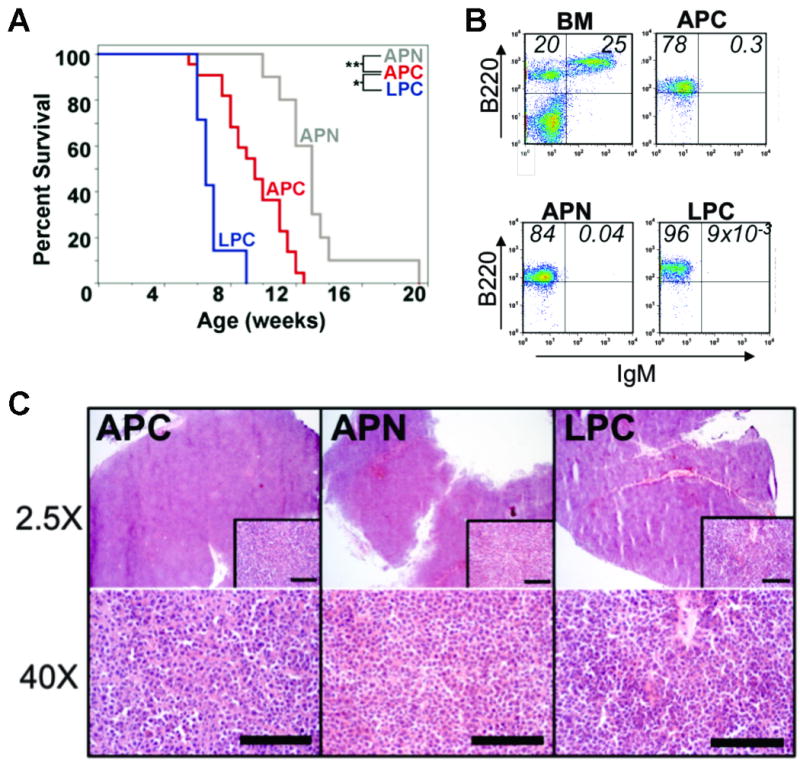
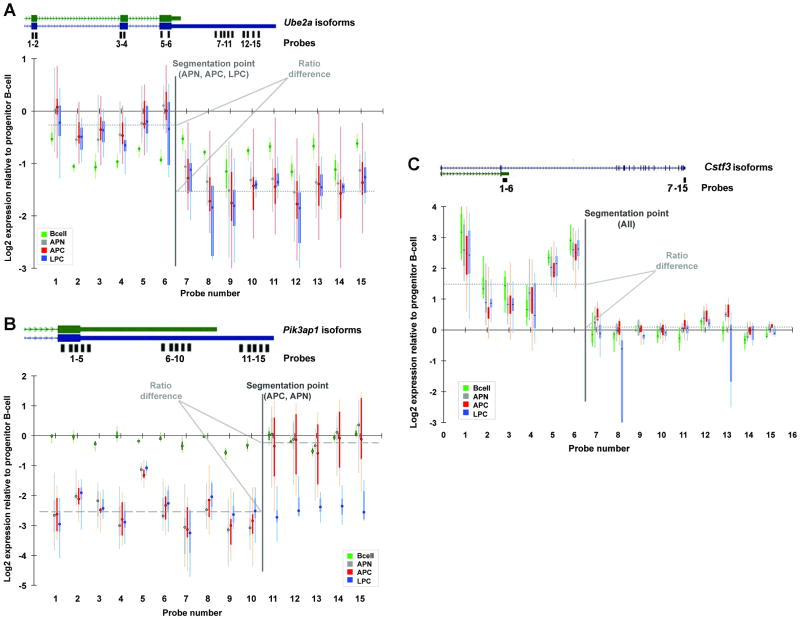
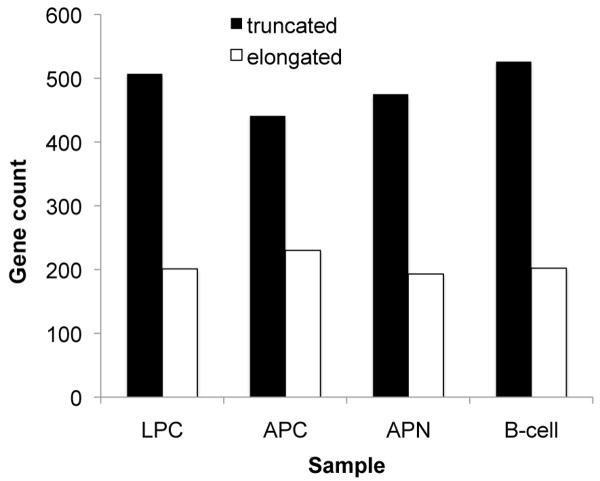
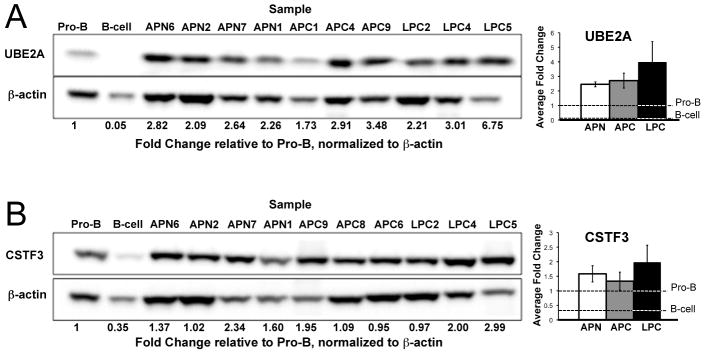
Molecular cancer diagnostics are an important clinical advance in cancer management, but new methods are still needed. In this context, gene expression signatures obtained by microarray represent a useful molecular diagnostic. Here, we describe novel probe-level microarray analyses that reveal connections between mRNA processing and neoplasia in multiple tumor types, with diagnostic potential. We now show that characteristic differences in mRNA processing, primarily in the 3'-untranslated region, define molecular signatures that can distinguish similar tumor subtypes with different survival characteristics, with at least 74% accuracy. Using a mouse model of B-cell leukemia/lymphoma, we find that differences in transcript isoform abundance are likely due to both alternative polyadenylation (APA) and differential degradation. While truncation of the 3'-UTR is the most common observed pattern, genes with elongated transcripts were also observed, and distinct groups of affected genes are found in related but distinct tumor types. Genes with elongated transcripts are overrepresented in ontology categories related to cell-cell adhesion and morphology. Analysis of microarray data from human primary tumor samples revealed similar phenomena. Western blot analysis of selected proteins confirms that changes in the 3'-UTR can correlate with changes in protein expression. Our work suggests that alternative mRNA processing, particularly APA, can be a powerful molecular biomarker with prognostic potential. Finally, these findings provide insights into the molecular mechanisms of gene deregulation in tumorigenesis.
Figures

References
-
- Bullinger L, Valk PJ. Gene expression profiling in acute myeloid leukemia. J Clin Oncol. 2005;23:6296–305. - PubMed
-
- Mano H. Stratification of acute myeloid leukemia based on gene expression profiles. Int J Hematol. 2004;80:389–94. - PubMed
-
- Mischel PS, Cloughesy TF, Nelson SF. DNA-microarray analysis of brain cancer: molecular classification for therapy. Nat Rev Neurosci. 2004;5:782–92. - PubMed
-
- Perez-Diez A, Morgun A, Shulzhenko N. Microarrays for cancer diagnosis and classification. Adv Exp Med Biol. 2007;593:74–85. - PubMed
-
- Kwan T, Benovoy D, Dias C, et al. Genome-wide analysis of transcript isoform variation in humans. Nat Genet. 2008;40:225–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
