

Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort
- PMID: 19937601
- PMCID: PMC3404892
- DOI: 10.1002/humu.21114
Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort
Abstract
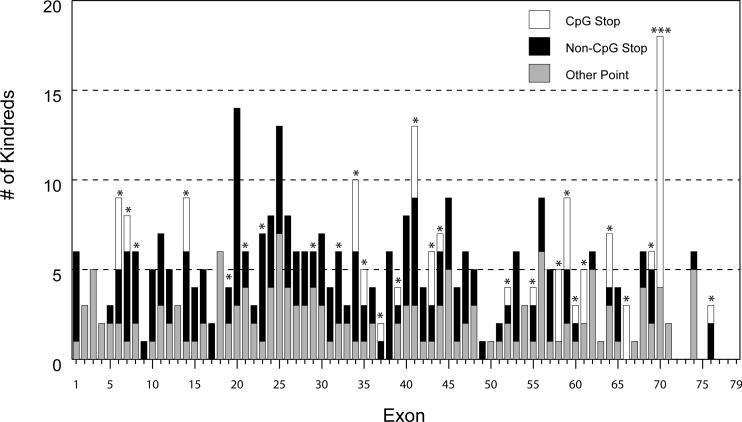
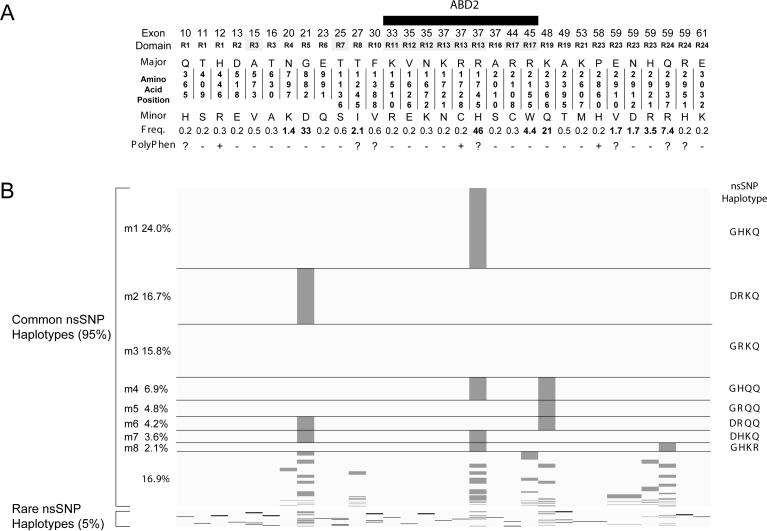
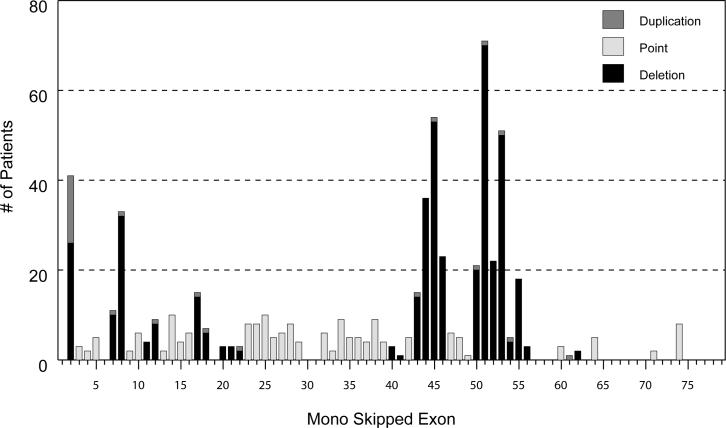
Mutations in the DMD gene, encoding the dystrophin protein, are responsible for the dystrophinopathies Duchenne Muscular Dystrophy (DMD), Becker Muscular Dystrophy (BMD), and X-linked Dilated Cardiomyopathy (XLDC). Mutation analysis has traditionally been challenging, due to the large gene size (79 exons over 2.2 Mb of genomic DNA). We report a very large aggregate data set comprised of DMD mutations detected in samples from patients enrolled in the United Dystrophinopathy Project, a multicenter research consortium, and in referral samples submitted for mutation analysis with a diagnosis of dystrophinopathy. We report 1,111 mutations in the DMD gene, including 891 mutations with associated phenotypes. These results encompass 506 point mutations (including 294 nonsense mutations) and significantly expand the number of mutations associated with the dystrophinopathies, highlighting the utility of modern diagnostic techniques. Our data supports the uniform hypermutability of CGA>TGA mutations, establishes the frequency of polymorphic muscle (Dp427m) protein isoforms and reveals unique genomic haplotypes associated with "private" mutations. We note that 60% of these patients would be predicted to benefit from skipping of a single DMD exon using antisense oligonucleotide therapy, and 62% would be predicted to benefit from an inclusive multiexonskipping approach directed toward exons 45 through 55.
Figures
References
-
- Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ, den Dunnen JT. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30(3):293–9. - PubMed
-
- Aartsma-Rus A, Janson AA, Kaman WE, Bremmer-Bout M, den Dunnen JT, Baas F, van Ommen GJ, van Deutekom JC. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum Mol Genet. 2003;12(8):907–14. - PubMed
-
- Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34(2):135–44. - PubMed
-
- Antonarakis SE, Krawczak M, Cooper DN. Disease-causing mutations in the human genome. Eur J Pediatr. 2000;159(Suppl 3):S173–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
