

Catalytic mechanism of human alpha-galactosidase
- PMID: 19940122
- PMCID: PMC2823503
- DOI: 10.1074/jbc.M109.060145
Catalytic mechanism of human alpha-galactosidase
Abstract
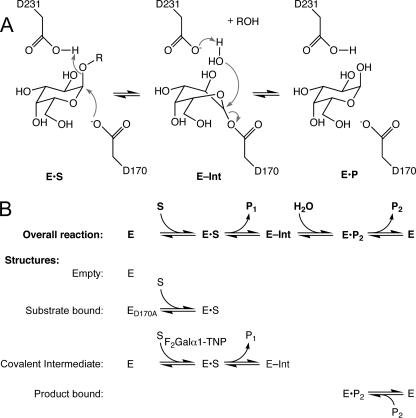
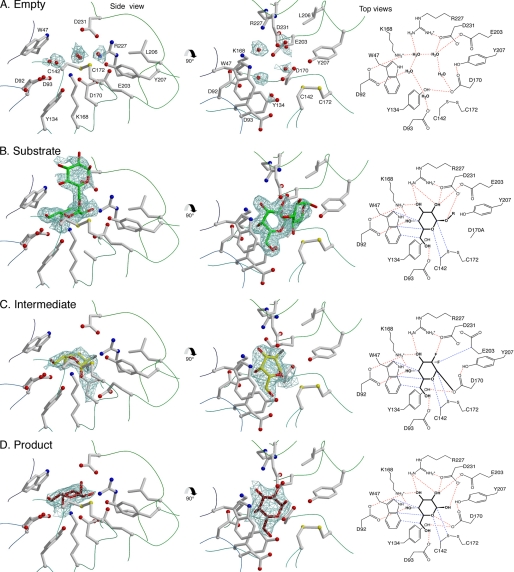
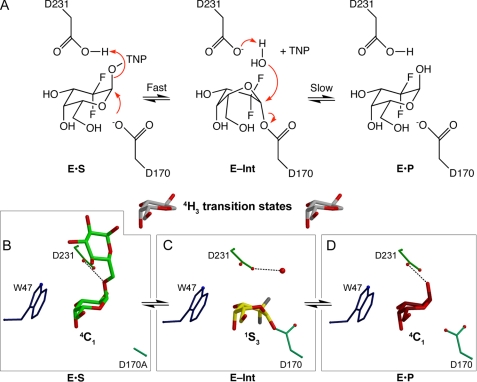
The enzyme alpha-galactosidase (alpha-GAL, also known as alpha-GAL A; E.C. 3.2.1.22) is responsible for the breakdown of alpha-galactosides in the lysosome. Defects in human alpha-GAL lead to the development of Fabry disease, a lysosomal storage disorder characterized by the buildup of alpha-galactosylated substrates in the tissues. alpha-GAL is an active target of clinical research: there are currently two treatment options for Fabry disease, recombinant enzyme replacement therapy (approved in the United States in 2003) and pharmacological chaperone therapy (currently in clinical trials). Previously, we have reported the structure of human alpha-GAL, which revealed the overall structure of the enzyme and established the locations of hundreds of mutations that lead to the development of Fabry disease. Here, we describe the catalytic mechanism of the enzyme derived from x-ray crystal structures of each of the four stages of the double displacement reaction mechanism. Use of a difluoro-alpha-galactopyranoside allowed trapping of a covalent intermediate. The ensemble of structures reveals distortion of the ligand into a (1)S(3) skew (or twist) boat conformation in the middle of the reaction cycle. The high resolution structures of each step in the catalytic cycle will allow for improved drug design efforts on alpha-GAL and other glycoside hydrolase family 27 enzymes by developing ligands that specifically target different states of the catalytic cycle. Additionally, the structures revealed a second ligand-binding site suitable for targeting by novel pharmacological chaperones.
Figures

Similar articles
-
Structural basis of Fabry disease.Mol Genet Metab. 2002 Sep-Oct;77(1-2):3-11. doi: 10.1016/s1096-7192(02)00151-8. Mol Genet Metab. 2002. PMID: 12359124 Review.
-
The molecular defect leading to Fabry disease: structure of human alpha-galactosidase.J Mol Biol. 2004 Mar 19;337(2):319-35. doi: 10.1016/j.jmb.2004.01.035. J Mol Biol. 2004. PMID: 15003450
-
Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin.Biochem J. 2007 Sep 1;406(2):285-95. doi: 10.1042/BJ20070479. Biochem J. 2007. PMID: 17555407 Free PMC article.
-
Fabry disease: characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele.Hum Mutat. 2003 Dec;22(6):486-92. doi: 10.1002/humu.10275. Hum Mutat. 2003. PMID: 14635108
-
Human α-Galactosidase A Mutants: Priceless Tools to Develop Novel Therapies for Fabry Disease.Int J Mol Sci. 2021 Jun 17;22(12):6518. doi: 10.3390/ijms22126518. Int J Mol Sci. 2021. PMID: 34204583 Free PMC article. Review.
Cited by
-
Mechanism-Based Allylic Carbasugar Chlorides That Form Covalent Intermediates with α- and β-Galactosidases.Molecules. 2024 Oct 14;29(20):4870. doi: 10.3390/molecules29204870. Molecules. 2024. PMID: 39459237 Free PMC article.
-
Thiogalactopyranosides are resistant to hydrolysis by α-galactosidases.Chembiochem. 2012 Jul 23;13(11):1673-9. doi: 10.1002/cbic.201200155. Epub 2012 Jun 27. Chembiochem. 2012. PMID: 22740420 Free PMC article.
-
Therapeutic Approaches in Lysosomal Storage Diseases.Biomolecules. 2021 Nov 26;11(12):1775. doi: 10.3390/biom11121775. Biomolecules. 2021. PMID: 34944420 Free PMC article. Review.
-
The pathogenesis of cerebral small vessel disease and vascular cognitive impairment.Physiol Rev. 2025 Jul 1;105(3):1075-1171. doi: 10.1152/physrev.00028.2024. Epub 2025 Feb 18. Physiol Rev. 2025. PMID: 39965059 Free PMC article. Review.
-
Biochemical Mechanisms beyond Glycosphingolipid Accumulation in Fabry Disease: Might They Provide Additional Therapeutic Treatments?J Clin Med. 2023 Mar 6;12(5):2063. doi: 10.3390/jcm12052063. J Clin Med. 2023. PMID: 36902850 Free PMC article. Review.
References
-
- Desnick R. J., Ioannou Y. A., Eng C. M. (2001) in The Metabolic and Molecular Bases of Inherited Disease (Scriver C. R., Beaudet A. L., Sly W. S., Valle D. eds), pp. 3733–3774, 8th Ed., McGraw-Hill, New York
-
- Liu Q. P., Sulzenbacher G., Yuan H., Bennett E. P., Pietz G., Saunders K., Spence J., Nudelman E., Levery S. B., White T., Neveu J. M., Lane W. S., Bourne Y., Olsson M. L., Henrissat B., Clausen H. (2007) Nat. Biotechnol. 25, 454–464 - PubMed
-
- Olsson M. L., Clausen H. (2008) Br. J. Haematol. 140, 3–12 - PubMed
-
- Brady R. O. (2003) Acta Paediatr. Suppl. 92, 19–24 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
