

Paucity of skeletal manifestations in Hispanic families with FBN1 mutations
- PMID: 19941982
- PMCID: PMC4354948
- DOI: 10.1016/j.ejmg.2009.11.001
Paucity of skeletal manifestations in Hispanic families with FBN1 mutations
Abstract
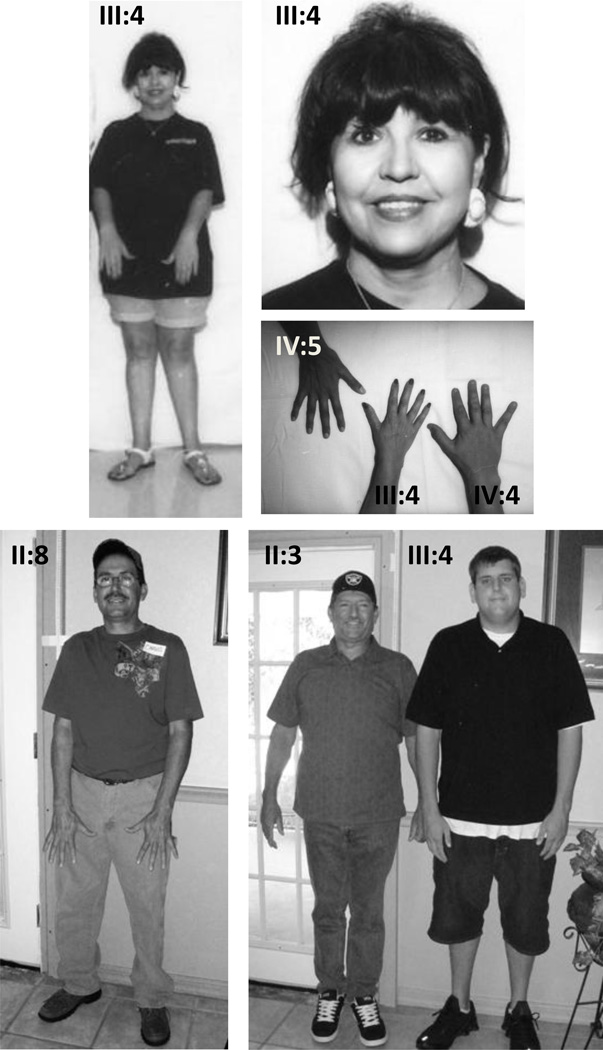
Marfan syndrome (MFS) is an autosomal dominant condition with pleiotropic manifestations involving the skeletal, ocular, and cardiovascular systems. The diagnosis is based primarily on clinical involvement of these and other systems, referred to as the Ghent criteria. We have identified three Hispanic families from Mexico with cardiovascular and ocular manifestations due to novel FBN1 mutations but with paucity of skeletal features. The largest family, hMFS001, had a frameshift mutation in exon 24 (3075delC) identified as the cause of aortic disease in the family. Assessment of eight affected adults revealed no major skeletal manifestation of MFS. Family hMFS002 had a missense mutation (R1530C) in exon 37. Four members fulfilled the criteria for ocular and cardiovascular phenotype but lacked skeletal manifestations. Family hMFS003 had two consecutive missense FBN1 mutations (C515W and R516G) in exon 12. Eight members fulfilled the ocular criteria for MFS and two members had major cardiovascular manifestations, however none of them met criteria for skeletal system. These data suggest that individuals of Hispanic descent with FBN1 mutations may not manifest skeletal features of the MFS to the same extent as Caucasians. We recommend that echocardiogram, ocular examination and FBN1 molecular testing be considered for any patients with possible MFS even in the absence of skeletal features, including Hispanic patients.
Copyright 2009 Elsevier Masson SAS. All rights reserved.
Figures
References
-
- Pyeritz RE. The Marfan syndrome. Annu. Rev. Med. 2000;51:481–510. - PubMed
-
- Pereira L, Levran O, Ramirez F, Lynch JR, Sykes B, Pyeritz RE, Dietz HC. A molecular approach to the stratification of cardiovascular risk in families with Marfan's syndrome [see comments] N. Engl. J. Med. 1994;331:148–153. - PubMed
-
- Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum. Mol. Genet. 1995;4(Spec No):1799–1809. - PubMed
-
- Ramirez F. Fibrillln mutations in Marfan syndrome and related phenotypes. Curr. Opin. Genet. Dev. 1996;6:309–315. - PubMed
-
- De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am. J. Med. Genet. 1996;62:417–426. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
