

Overexpression of RAD51 suppresses recombination defects: a possible mechanism to reverse genomic instability
- PMID: 19942681
- PMCID: PMC2831301
- DOI: 10.1093/nar/gkp1063
Overexpression of RAD51 suppresses recombination defects: a possible mechanism to reverse genomic instability
Abstract
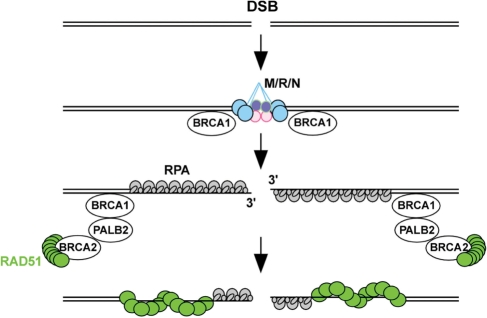
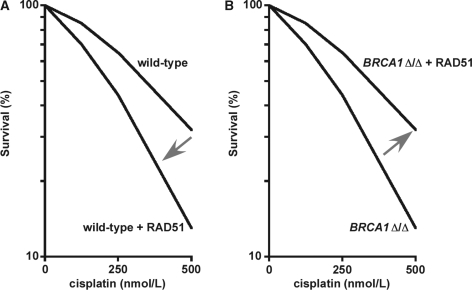
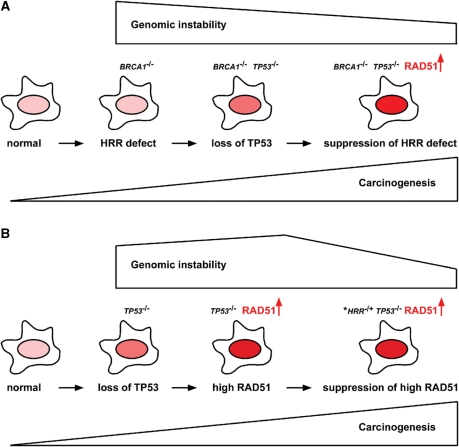
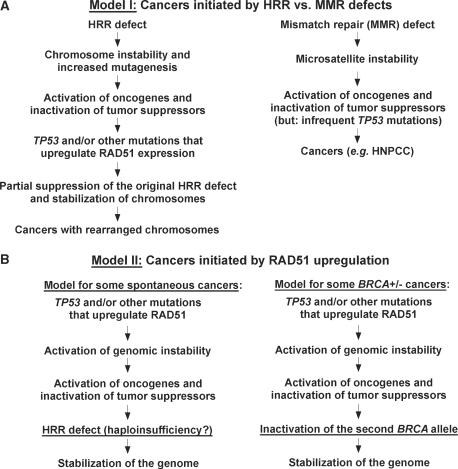
RAD51, a key protein in the homologous recombinational DNA repair (HRR) pathway, is the major strand-transferase required for mitotic recombination. An important early step in HRR is the formation of single-stranded DNA (ss-DNA) coated by RPA (a ss-DNA-binding protein). Displacement of RPA by RAD51 is highly regulated and facilitated by a number of different proteins known as the 'recombination mediators'. To assist these recombination mediators, a second group of proteins also is required and we are defining these proteins here as 'recombination co-mediators'. Defects in either recombination mediators or co-mediators, including BRCA1 and BRCA2, lead to impaired HRR that can genetically be complemented for (i.e. suppressed) by overexpression of RAD51. Defects in HRR have long been known to contribute to genomic instability leading to tumor development. Since genomic instability also slows cell growth, precancerous cells presumably require genomic re-stabilization to gain a growth advantage. RAD51 is overexpressed in many tumors, and therefore, we hypothesize that the complementing ability of elevated levels of RAD51 in tumors with initial HRR defects limits genomic instability during carcinogenic progression. Of particular interest, this model may also help explain the high frequency of TP53 mutations in human cancers, since wild-type p53 represses RAD51 expression.
Figures
Similar articles
-
Inactivation of checkpoint kinase 1 (Chk1) during parvovirus minute virus of mice (MVM) infection inhibits cellular homologous recombination repair and facilitates viral genome replication.J Virol. 2024 Dec 17;98(12):e0088924. doi: 10.1128/jvi.00889-24. Epub 2024 Nov 20. J Virol. 2024. PMID: 39565136 Free PMC article.
-
Methotrexate-mediated inhibition of RAD51 expression and homologous recombination in cancer cells.J Cancer Res Clin Oncol. 2012 May;138(5):811-8. doi: 10.1007/s00432-011-1132-8. Epub 2012 Jan 25. J Cancer Res Clin Oncol. 2012. PMID: 22274865 Free PMC article.
-
ZNF280A links DNA double-strand break repair to human 22q11.2 distal deletion syndrome.Nat Cell Biol. 2025 Jun;27(6):1006-1020. doi: 10.1038/s41556-025-01674-1. Epub 2025 Jun 16. Nat Cell Biol. 2025. PMID: 40523937
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
-
The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review.Int J Mol Sci. 2022 Jul 23;23(15):8125. doi: 10.3390/ijms23158125. Int J Mol Sci. 2022. PMID: 35897700 Free PMC article.
Cited by
-
The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers.Mol Cell. 2019 Jan 17;73(2):224-237.e6. doi: 10.1016/j.molcel.2018.11.003. Epub 2018 Dec 13. Mol Cell. 2019. PMID: 30554948 Free PMC article.
-
SOSSB1 and SOSSB2 mutually regulate protein stability through competitive binding of SOSSA.Cell Death Discov. 2023 Aug 28;9(1):319. doi: 10.1038/s41420-023-01619-3. Cell Death Discov. 2023. PMID: 37640700 Free PMC article.
-
Loss of heterozygosity for chromosomal regions 15q14-21.1, 17q21.31, and 13q12.3-13.1 and its relevance for prostate cancer.Med Oncol. 2015 Nov;32(11):246. doi: 10.1007/s12032-015-0691-y. Epub 2015 Oct 3. Med Oncol. 2015. PMID: 26433958 Free PMC article.
-
β1-Integrin Impacts Rad51 Stability and DNA Double-Strand Break Repair by Homologous Recombination.Mol Cell Biol. 2018 Apr 16;38(9):e00672-17. doi: 10.1128/MCB.00672-17. Print 2018 May 1. Mol Cell Biol. 2018. PMID: 29463647 Free PMC article.
-
DNA repair pathways in trypanosomatids: from DNA repair to drug resistance.Microbiol Mol Biol Rev. 2014 Mar;78(1):40-73. doi: 10.1128/MMBR.00045-13. Microbiol Mol Biol Rev. 2014. PMID: 24600040 Free PMC article. Review.
References
-
- Venkitaraman AR. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Ann. Rev. Pathol. 2009;4:461–487. - PubMed
-
- Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–105. - PubMed
-
- Ralhan R, Kaur J, Kreienberg R, Wiesmuller L. Links between DNA double strand break repair and breast cancer: accumulating evidence from both familial and nonfamilial cases. Cancer Lett. 2007;248:1–17. - PubMed
-
- Gatz SA, Wiesmuller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–1016. - PubMed
-
- Lazaro-Trueba I, Arias C, Silva A. Double bolt regulation of Rad51 by p53: a role for transcriptional repression. Cell Cycle. 2006;5:1062–1065. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
