

PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis
- PMID: 19942859
- PMCID: PMC2824464
- DOI: 10.1038/emboj.2009.339
PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis
Abstract
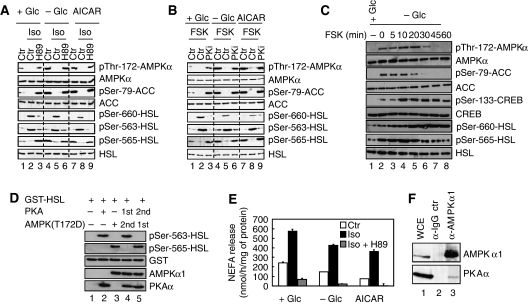
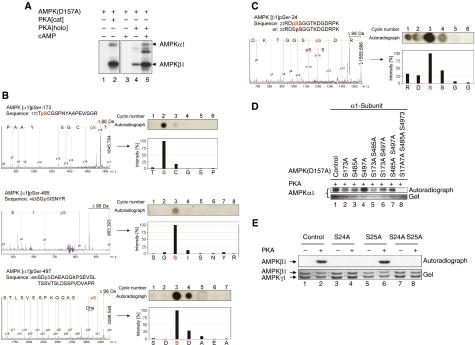
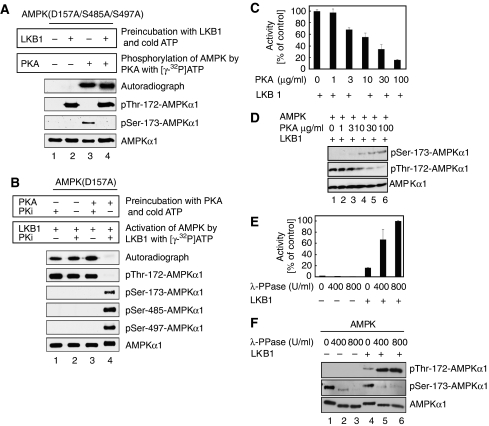
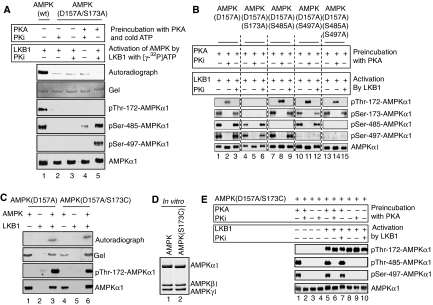
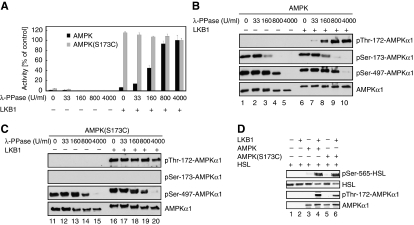
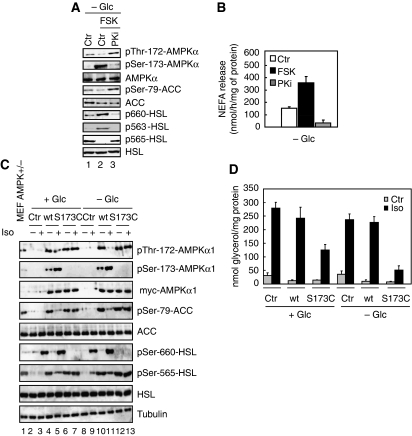
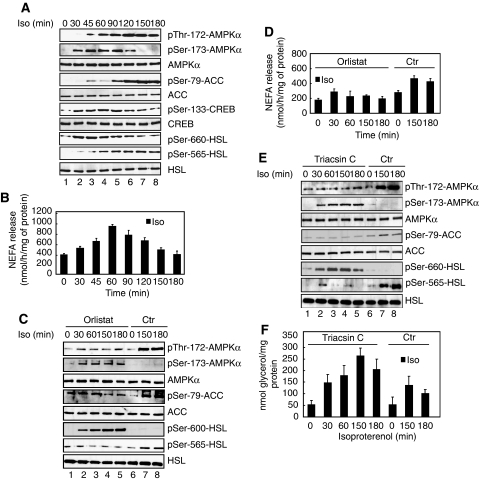
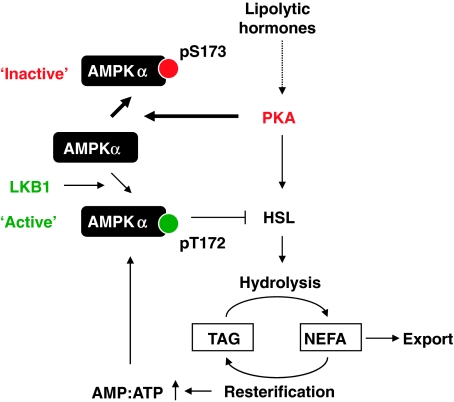
The mobilization of metabolic energy from adipocytes depends on a tightly regulated balance between hydrolysis and resynthesis of triacylglycerides (TAGs). Hydrolysis is stimulated by beta-adrenergic signalling to PKA that mediates phosphorylation of lipolytic enzymes, including hormone-sensitive lipase (HSL). TAG resynthesis is associated with high-energy consumption, which when inordinate, leads to increased AMPK activity that acts to restrain hydrolysis of TAGs by inhibiting PKA-mediated activation of HSL. Here, we report that in primary mouse adipocytes, PKA associates with and phosphorylates AMPKalpha1 at Ser-173 to impede threonine (Thr-172) phosphorylation and thus activation of AMPKalpha1 by LKB1 in response to lipolytic signals. Activation of AMPKalpha1 by LKB1 is also blocked by PKA-mediated phosphorylation of AMPKalpha1 in vitro. Functional analysis of an AMPKalpha1 species carrying a non-phosphorylatable mutation at Ser-173 revealed a critical function of this phosphorylation for efficient release of free fatty acids and glycerol in response to PKA-activating signals. These results suggest a new mechanism of negative regulation of AMPK activity by PKA that is important for converting a lipolytic signal into an effective lipolytic response.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Synergistic effects of cAMP-dependent protein kinase A and AMP-activated protein kinase on lipolysis in kinsenoside-treated C3H10T1/2 adipocytes.Phytomedicine. 2019 Mar 1;55:255-263. doi: 10.1016/j.phymed.2018.06.043. Epub 2018 Aug 11. Phytomedicine. 2019. PMID: 30668437
-
Mechanisms of metformin inhibiting lipolytic response to isoproterenol in primary rat adipocytes.J Mol Endocrinol. 2009 Jan;42(1):57-66. doi: 10.1677/JME-08-0130. Epub 2008 Oct 27. J Mol Endocrinol. 2009. PMID: 18955435
-
Reduced ATGL-mediated lipolysis attenuates β-adrenergic-induced AMPK signaling, but not the induction of PKA-targeted genes, in adipocytes and adipose tissue.Am J Physiol Cell Physiol. 2016 Aug 1;311(2):C269-76. doi: 10.1152/ajpcell.00126.2016. Epub 2016 Jun 29. Am J Physiol Cell Physiol. 2016. PMID: 27357546 Free PMC article.
-
mTORC1 inhibition via rapamycin promotes triacylglycerol lipolysis and release of free fatty acids in 3T3-L1 adipocytes.Lipids. 2010 Dec;45(12):1089-100. doi: 10.1007/s11745-010-3488-y. Epub 2010 Nov 2. Lipids. 2010. PMID: 21042876 Free PMC article.
-
Molecular mechanisms regulating hormone-sensitive lipase and lipolysis.Biochem Soc Trans. 2003 Dec;31(Pt 6):1120-4. doi: 10.1042/bst0311120. Biochem Soc Trans. 2003. PMID: 14641008 Review.
Cited by
-
Can UPR integrate fasting and stem cell regeneration?Front Chem. 2015 Feb 3;3:5. doi: 10.3389/fchem.2015.00005. eCollection 2015. Front Chem. 2015. PMID: 25692126 Free PMC article. No abstract available.
-
Anchored phosphatases modulate glucose homeostasis.EMBO J. 2012 Oct 17;31(20):3991-4004. doi: 10.1038/emboj.2012.244. Epub 2012 Aug 31. EMBO J. 2012. PMID: 22940692 Free PMC article.
-
Acyl-coenzyme A synthetases in metabolic control.Curr Opin Lipidol. 2010 Jun;21(3):212-7. doi: 10.1097/mol.0b013e32833884bb. Curr Opin Lipidol. 2010. PMID: 20480548 Free PMC article. Review.
-
The regulation of autophagy - unanswered questions.J Cell Sci. 2011 Jan 15;124(Pt 2):161-70. doi: 10.1242/jcs.064576. J Cell Sci. 2011. PMID: 21187343 Free PMC article. Review.
-
Frontier of epilepsy research - mTOR signaling pathway.Exp Mol Med. 2011 May 31;43(5):231-74. doi: 10.3858/emm.2011.43.5.032. Exp Mol Med. 2011. PMID: 21467839 Free PMC article. Review.
References
-
- Anthonsen MW, Ronnstrand L, Wernstedt C, Degerman E, Holm C (1998) Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem 273: 215–221 - PubMed
-
- Bergeron R, Previs SF, Cline GW, Perret P, Russell RR III, Young LH, Shulman GI (2001) Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes 50: 1076–1082 - PubMed
-
- Brooks B, Arch JR, Newsholme EA (1982) Effects of hormones on the rate of the triacylglycerol/fatty acid substrate cycle in adipocytes and epididymal fat pads. FEBS Lett 146: 327–330 - PubMed
-
- Carling D, Sanders MJ, Woods A (2008) The regulation of AMP-activated protein kinase by upstream kinases. Int J Obes (Lond) 32(Suppl 4): S55–S59 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
