

How many 3D structures do we need to train a predictor?
- PMID: 19944385
- PMCID: PMC5054404
- DOI: 10.1016/S1672-0229(08)60041-8
How many 3D structures do we need to train a predictor?
Abstract
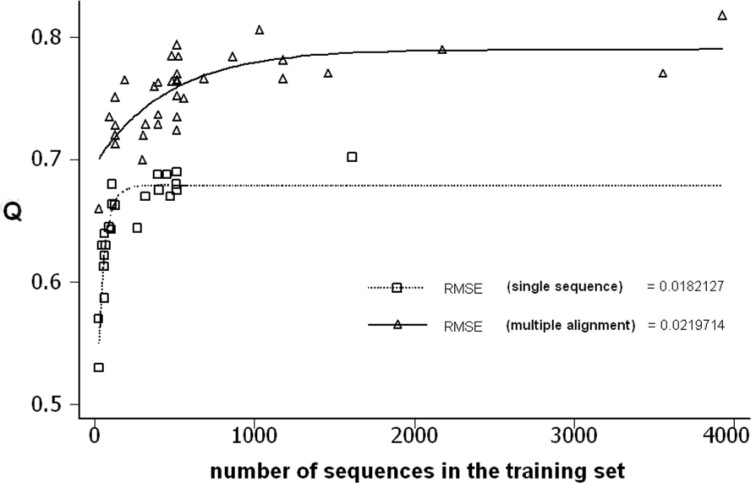
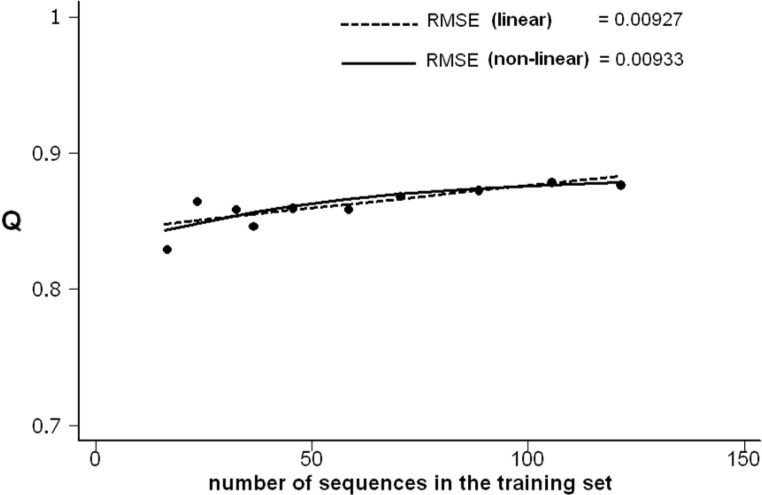
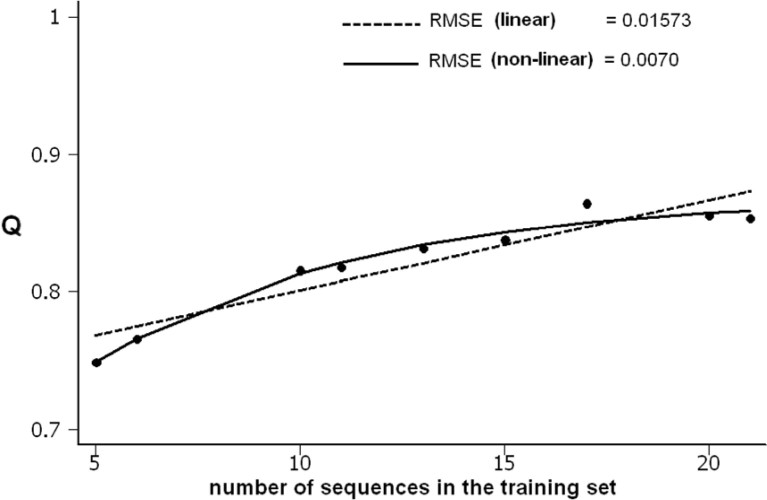
It has been shown that the progress in the determination of membrane protein structure grows exponentially, with approximately the same growth rate as that of the water-soluble proteins. In order to investigate the effect of this, on the performance of prediction algorithms for both alpha-helical and beta-barrel membrane proteins, we conducted a prospective study based on historical records. We trained separate hidden Markov models with different sized training sets and evaluated their performance on topology prediction for the two classes of transmembrane proteins. We show that the existing top-scoring algorithms for predicting the transmembrane segments of alpha-helical membrane proteins perform slightly better than that of beta-barrel outer membrane proteins in all measures of accuracy. With the same rationale, a meta-analysis of the performance of the secondary structure prediction algorithms indicates that existing algorithmic techniques cannot be further improved by just adding more non-homologous sequences to the training sets. The upper limit for secondary structure prediction is estimated to be no more than 70% and 80% of correctly predicted residues for single sequence based methods and multiple sequence based ones, respectively. Therefore, we should concentrate our efforts on utilizing new techniques for the development of even better scoring predictors.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
