

Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies
- PMID: 19946270
- PMCID: PMC4002225
- DOI: 10.1038/ng.491
Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies
Abstract
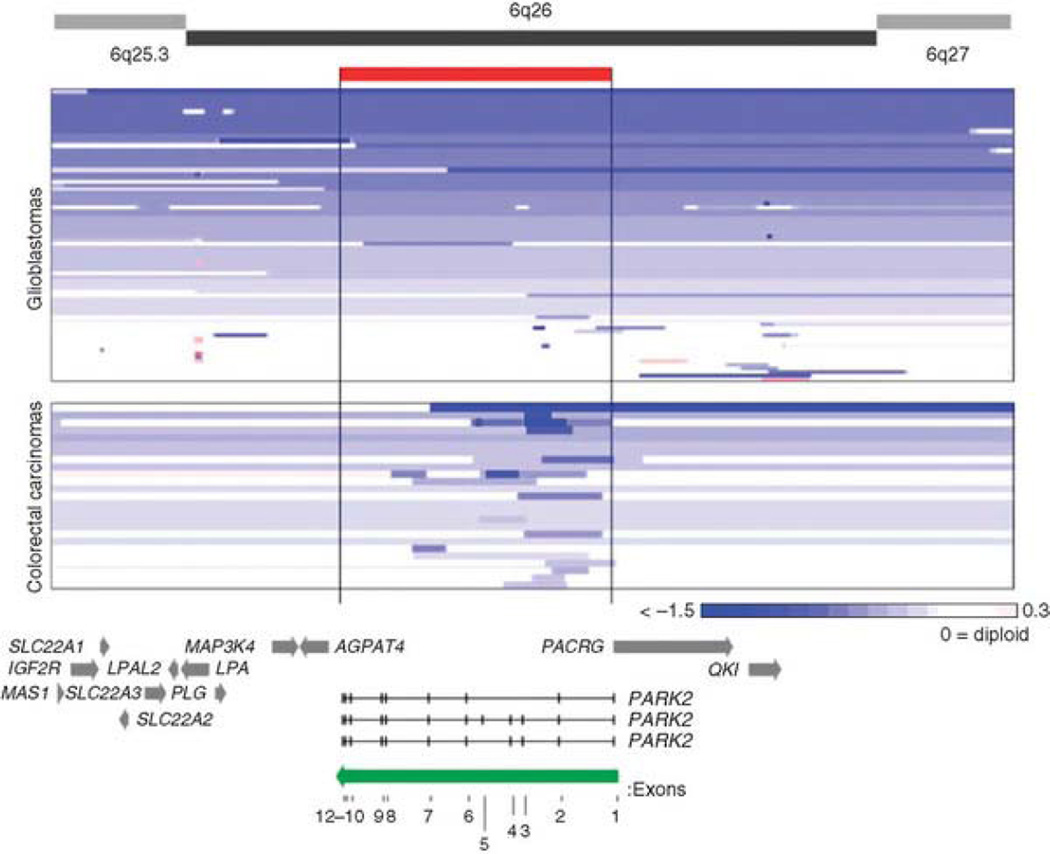
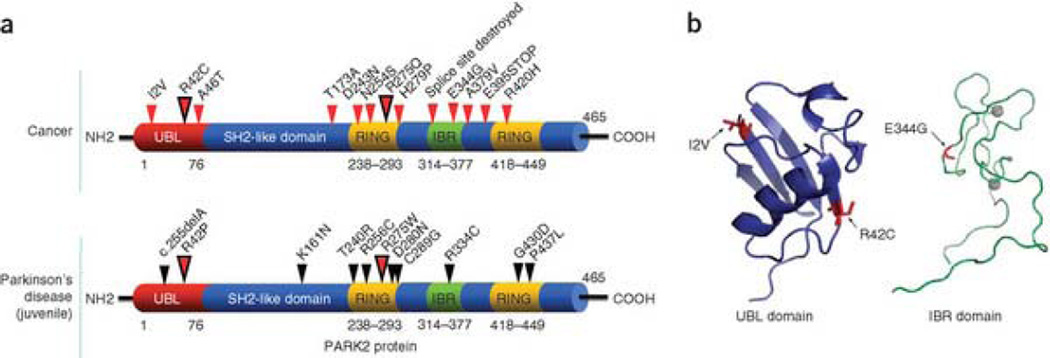
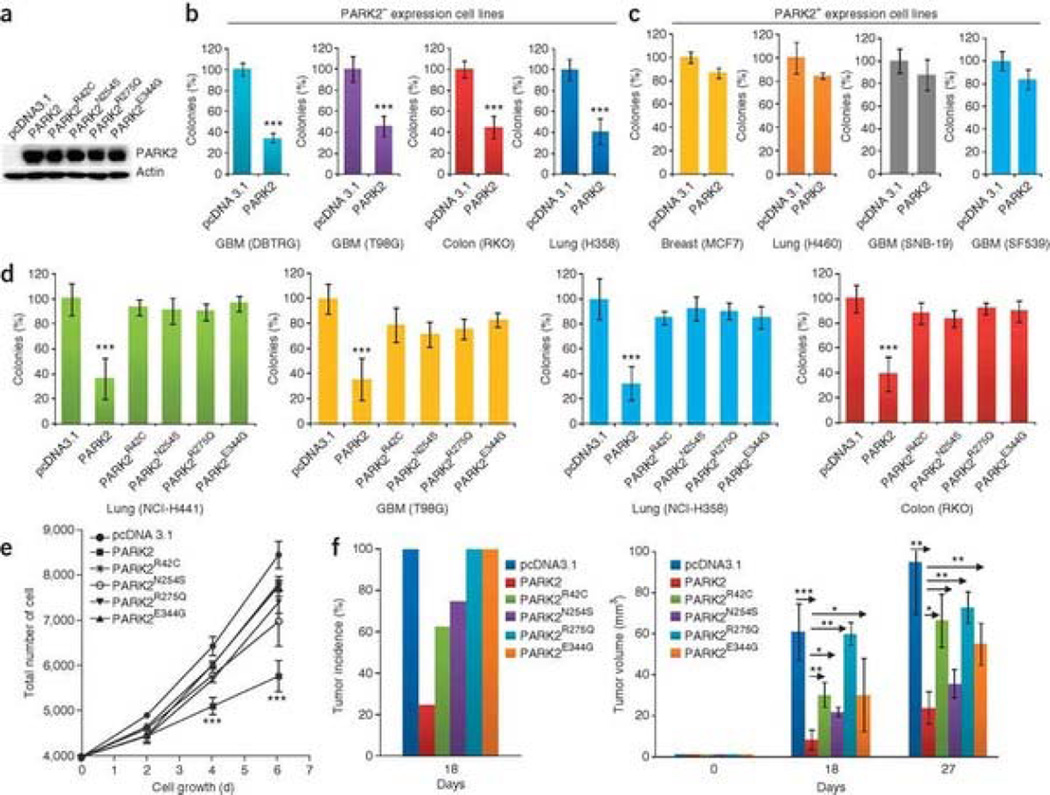
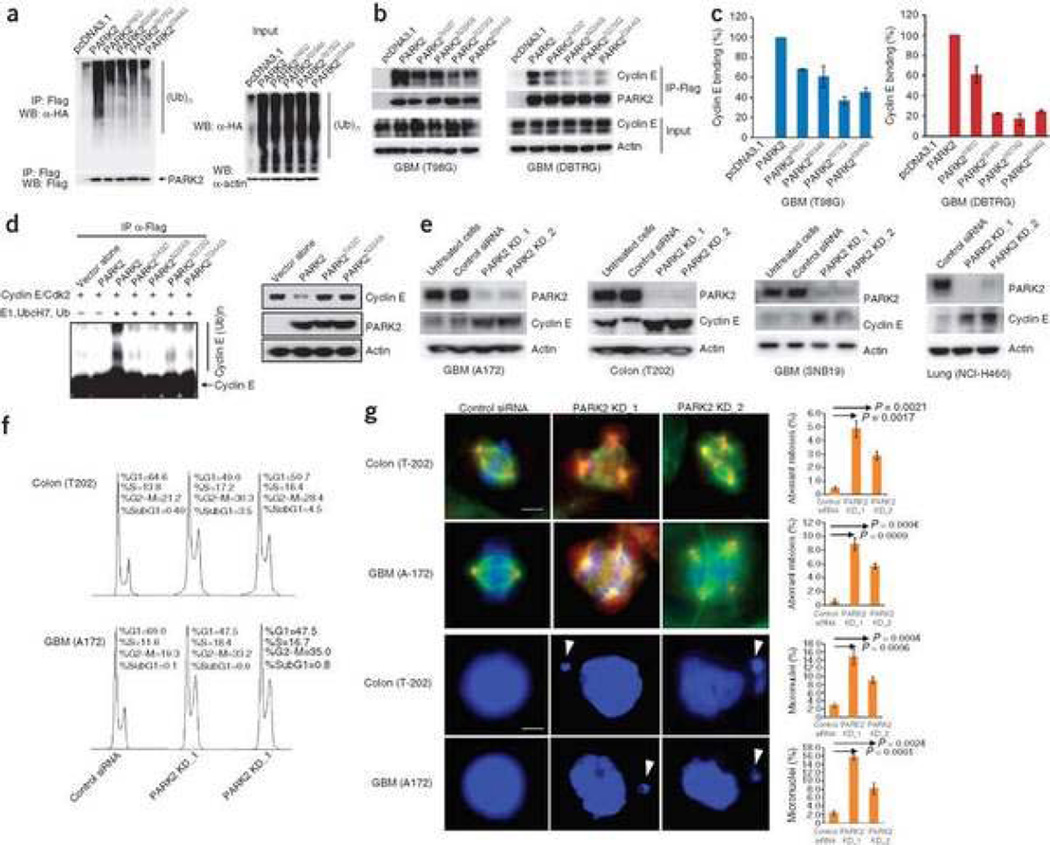
Mutation of the gene PARK2, which encodes an E3 ubiquitin ligase, is the most common cause of early-onset Parkinson's disease. In a search for multisite tumor suppressors, we identified PARK2 as a frequently targeted gene on chromosome 6q25.2-q27 in cancer. Here we describe inactivating somatic mutations and frequent intragenic deletions of PARK2 in human malignancies. The PARK2 mutations in cancer occur in the same domains, and sometimes at the same residues, as the germline mutations causing familial Parkinson's disease. Cancer-specific mutations abrogate the growth-suppressive effects of the PARK2 protein. PARK2 mutations in cancer decrease PARK2's E3 ligase activity, compromising its ability to ubiquitinate cyclin E and resulting in mitotic instability. These data strongly point to PARK2 as a tumor suppressor on 6q25.2-q27. Thus, PARK2, a gene that causes neuronal dysfunction when mutated in the germline, may instead contribute to oncogenesis when altered in non-neuronal somatic cells.
Figures
References
-
- Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. - PubMed
-
- Lücking CB, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N. Engl J. Med. 2000;342:1560–1567. - PubMed
-
- Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. - PubMed
-
- Samii A, Nutt JG, Ransom BR. Parkinson's disease. Lancet. 2004;363:1783–1793. - PubMed
-
- Abbas N, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum. Mol. Genet. 1999;8:567–574. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
