

Inducible priming phosphorylation promotes ligand-independent degradation of the IFNAR1 chain of type I interferon receptor
- PMID: 19948722
- PMCID: PMC2807289
- DOI: 10.1074/jbc.M109.071498
Inducible priming phosphorylation promotes ligand-independent degradation of the IFNAR1 chain of type I interferon receptor
Abstract
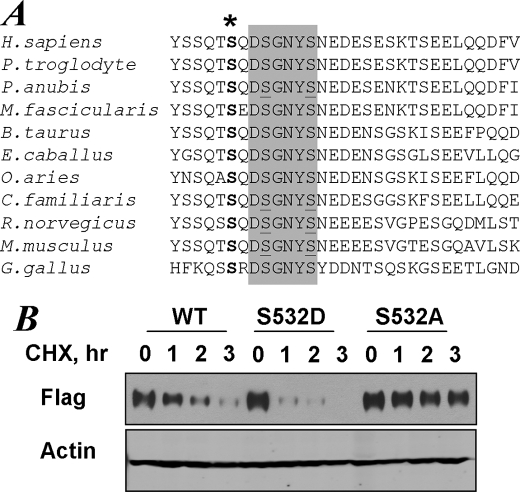
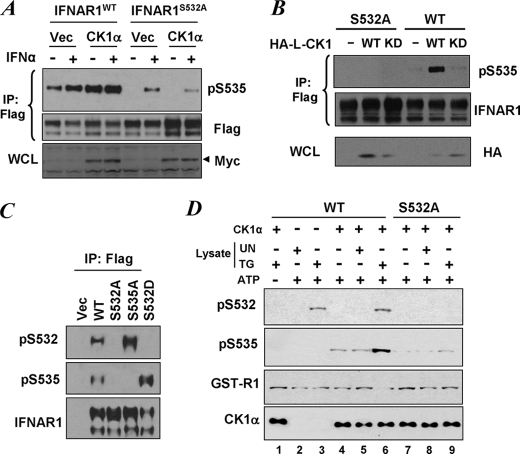
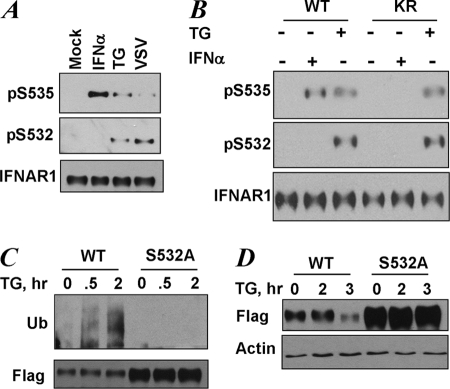
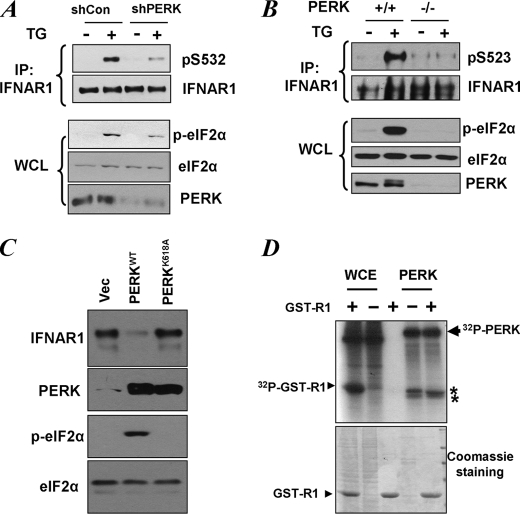
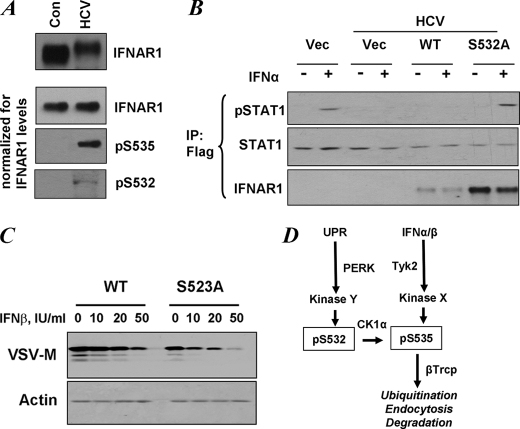
Phosphorylation-dependent ubiquitination and ensuing down-regulation and lysosomal degradation of the interferon alpha/beta receptor chain 1 (IFNAR1) of the receptor for Type I interferons play important roles in limiting the cellular responses to these cytokines. These events could be stimulated either by the ligands (in a Janus kinase-dependent manner) or by unfolded protein response (UPR) inducers including viral infection (in a manner dependent on the activity of pancreatic endoplasmic reticulum kinase). Both ligand-dependent and -independent pathways converge on phosphorylation of Ser(535) within the IFNAR1 degron leading to recruitment of beta-Trcp E3 ubiquitin ligase and concomitant ubiquitination and degradation. Casein kinase 1 alpha (CK1 alpha) was shown to directly phosphorylate Ser(535) within the ligand-independent pathway. Yet given the constitutive activity of CK1 alpha, it remained unclear how this pathway is stimulated by UPR. Here we report that induction of UPR promotes the phosphorylation of a proximal residue, Ser(532), in a pancreatic endoplasmic reticulum kinase-dependent manner. This serine serves as a priming site that promotes subsequent phosphorylation of IFNAR1 within its degron by CK1 alpha. These events play an important role in regulating ubiquitination and degradation of IFNAR1 as well as the extent of Type I interferon signaling.
Figures
References
-
- Wallach D. F. (1987) Fundamentals of Receptor Molecular Biology, M. Dekker, New York
-
- Müller U., Steinhoff U., Reis L. F., Hemmi S., Pavlovic J., Zinkernagel R. M., Aguet M. (1994) Science. 264, 1918–1921 - PubMed
-
- Billard C., Sigaux F., Castaigne S., Valensi F., Flandrin G., Degos L., Falcoff E., Aguet M. (1986) Blood. 67, 821–826 - PubMed
-
- Maxwell B. L., Talpaz M., Gutterman J. U. (1985) Int. J. Cancer 36, 23–28 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
