

Genetic association tests: a method for the joint analysis of family and case-control data
- PMID: 19951892
- PMCID: PMC2874328
- DOI: 10.1186/1479-7364-4-1-2
Genetic association tests: a method for the joint analysis of family and case-control data
Abstract
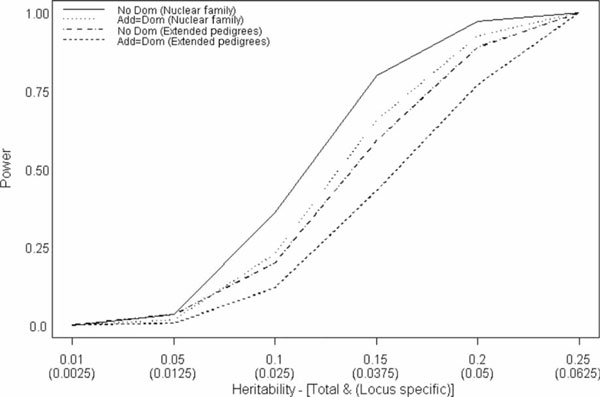
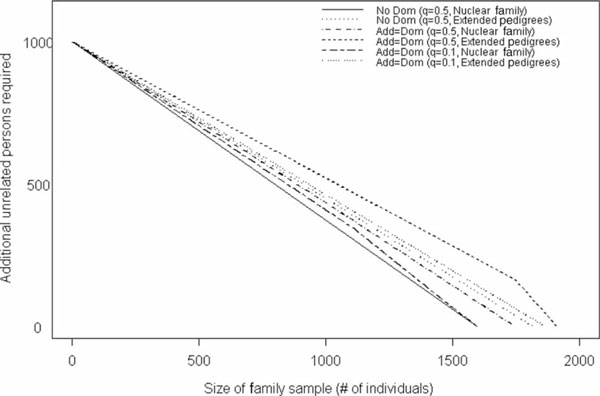
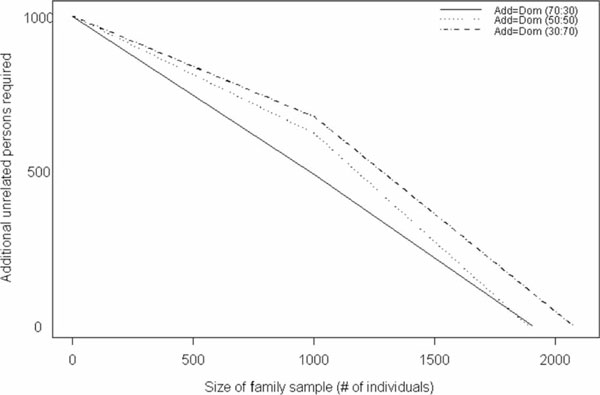
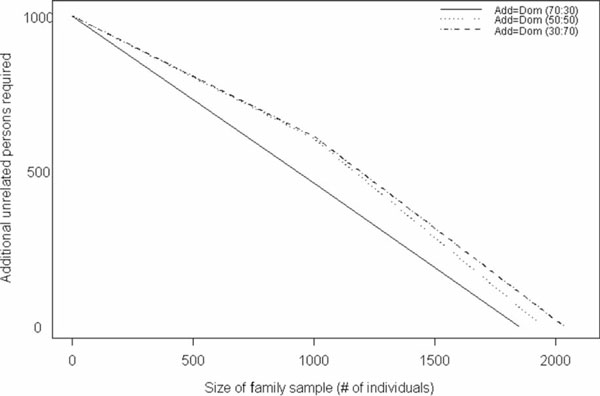
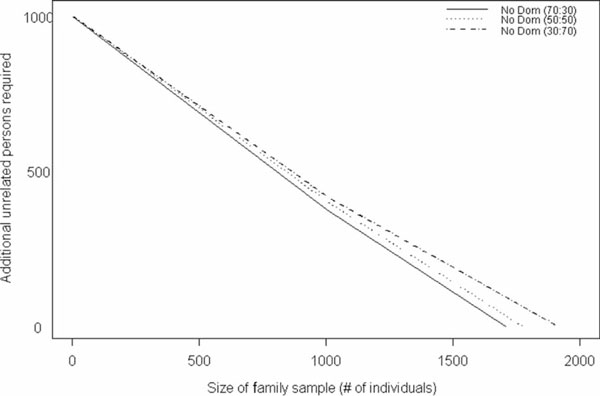
With the trend in molecular epidemiology towards both genome-wide association studies and complex modelling, the need for large sample sizes to detect small effects and to allow for the estimation of many parameters within a model continues to increase. Unfortunately, most methods of association analysis have been restricted to either a family-based or a case-control design, resulting in the lack of synthesis of data from multiple studies. Transmission disequilibrium-type methods for detecting linkage disequilibrium from family data were developed as an effective way of preventing the detection of association due to population stratification. Because these methods condition on parental genotype, however, they have precluded the joint analysis of family and case-control data, although methods for case-control data may not protect against population stratification and do not allow for familial correlations. We present here an extension of a family-based association analysis method for continuous traits that will simultaneously test for, and if necessary control for, population stratification. We further extend this method to analyse binary traits (and therefore family and case-control data together) and accurately to estimate genetic effects in the population, even when using an ascertained family sample. Finally, we present the power of this binary extension for both family-only and joint family and case-control data, and demonstrate the accuracy of the association parameter and variance components in an ascertained family sample.
Figures
References
-
- Elston RC. 'Linkage and association to genetic markers'. Exp Clin Immunogenet. 1995;12:129–140. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
