

Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction
- PMID: 19953085
- PMCID: PMC2824489
- DOI: 10.1038/msb.2009.87
Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction
Abstract
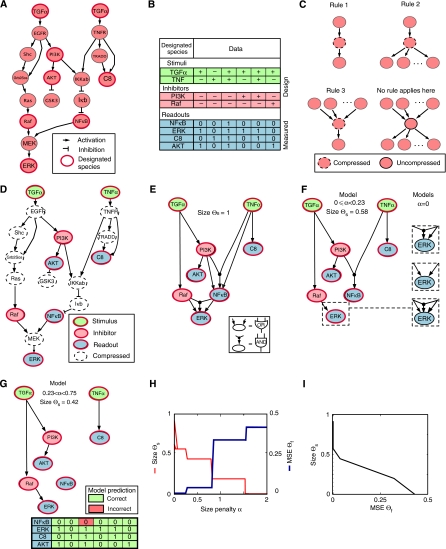
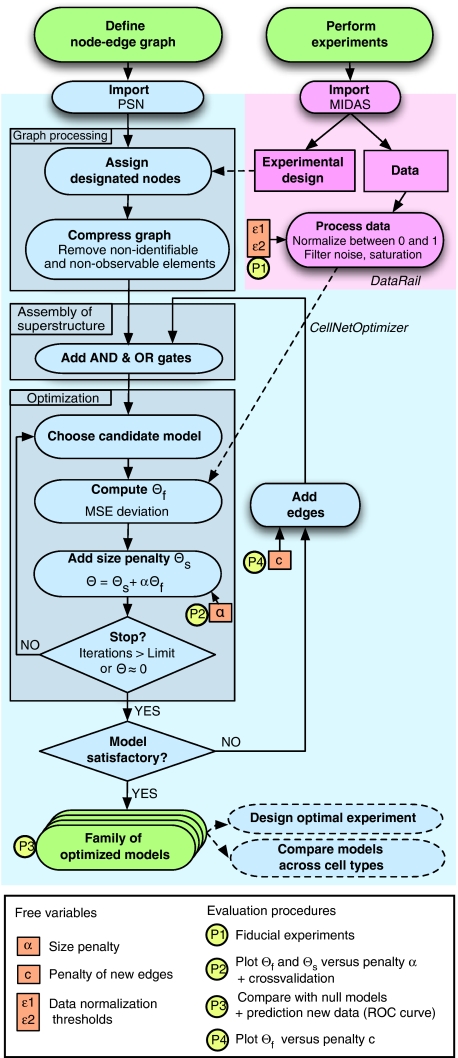
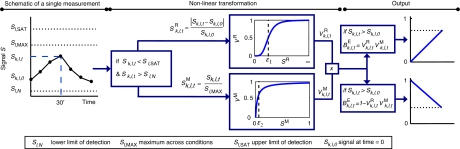
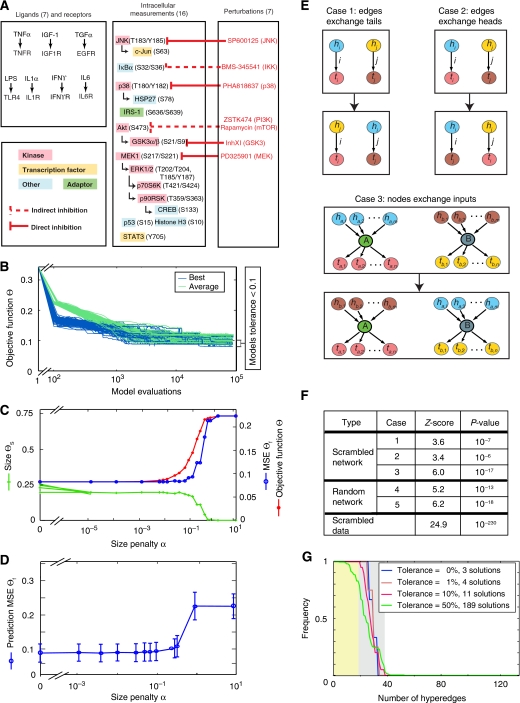
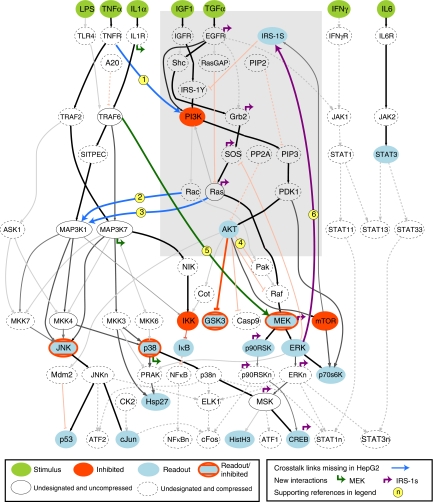
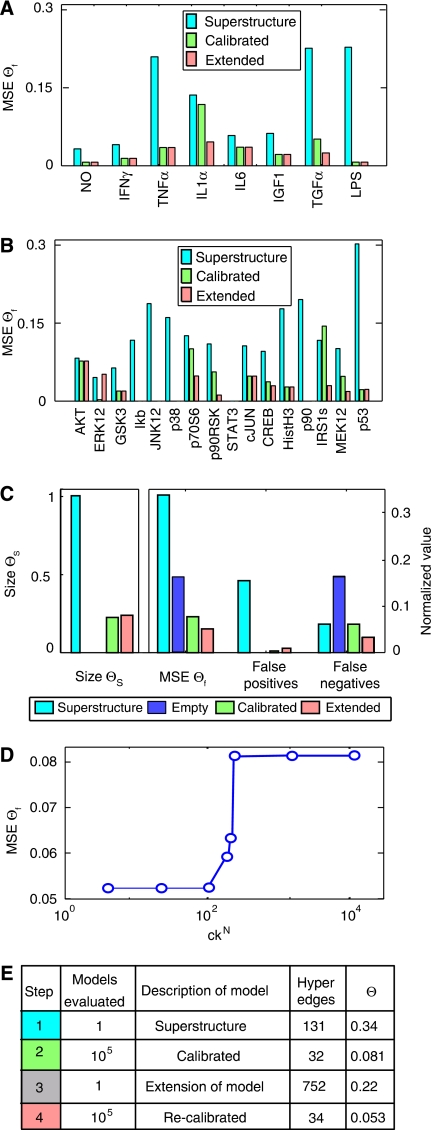
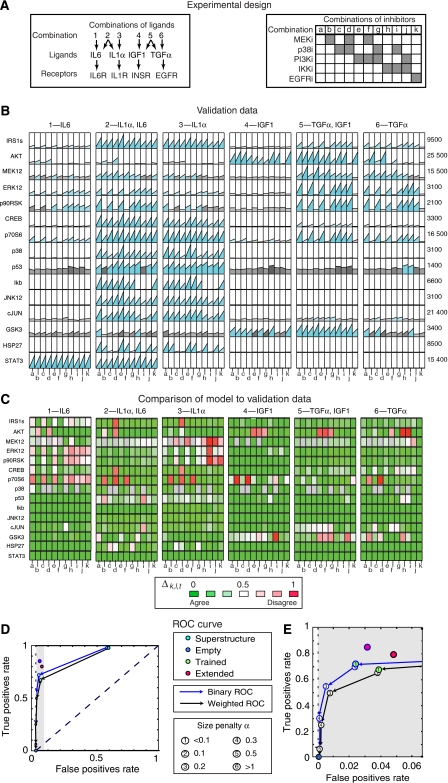
Large-scale protein signalling networks are useful for exploring complex biochemical pathways but do not reveal how pathways respond to specific stimuli. Such specificity is critical for understanding disease and designing drugs. Here we describe a computational approach--implemented in the free CNO software--for turning signalling networks into logical models and calibrating the models against experimental data. When a literature-derived network of 82 proteins covering the immediate-early responses of human cells to seven cytokines was modelled, we found that training against experimental data dramatically increased predictive power, despite the crudeness of Boolean approximations, while significantly reducing the number of interactions. Thus, many interactions in literature-derived networks do not appear to be functional in the liver cells from which we collected our data. At the same time, CNO identified several new interactions that improved the match of model to data. Although missing from the starting network, these interactions have literature support. Our approach, therefore, represents a means to generate predictive, cell-type-specific models of mammalian signalling from generic protein signalling networks.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Akaike H (1974) A new look at the statistical model identification. Automat Contr 19: 716–723
-
- Aldridge BB, Burke JM, Lauffenburger DA, Sorger PK (2006) Physicochemical modelling of cell signalling pathways. Nat Cell Biol 8: 1195–1203 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
