

Non-imprinted allele-specific DNA methylation on human autosomes
- PMID: 19958531
- PMCID: PMC2812945
- DOI: 10.1186/gb-2009-10-12-r138
Non-imprinted allele-specific DNA methylation on human autosomes
Abstract
Background: Differential DNA methylation between alleles is well established in imprinted genes and the X chromosomes in females but has rarely been reported at non-imprinted loci on autosomes.
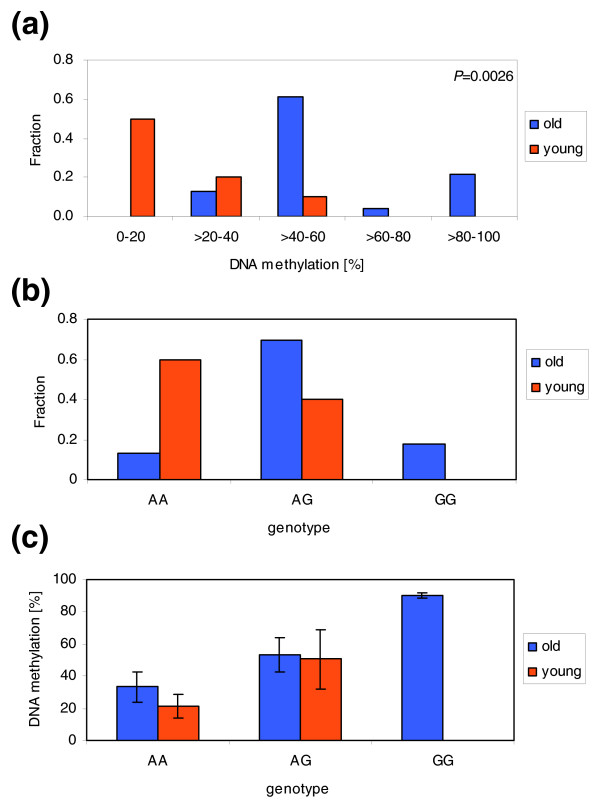
Results: We studied DNA methylation of cytosine-guanine dinucleotide (CpG) islands on chromosome 21 in leukocytes from several healthy individuals and observed novel cases of pronounced differential methylation of alleles. Allele-specific methylation affected complete CpG islands with methylation differences between alleles of up to 85%. The methylation differences between alleles were strongly correlated with the genotypes, excluding a connection to imprinting. We show that allele-specific methylation can lead to allelic repression of the methylated gene copy. Based on our results, allele-specific methylation is likely to affect about 10% of all human genes and to contribute to allele-specific expression and monoallelic gene silencing. Therefore, allele-specific methylation represents an epigenetic pathway of how genetic polymorphisms may lead to phenotypic variability. In most cases, we observed that some, but not all, heterozygous individuals showed allele-specific methylation, suggesting that allele-specific methylation is the outcome of an epigenetic drift, the direction of which is determined by the genetic differences between the alleles. We could show that the tendency to acquire hypermethylation in one allele was inherited.
Conclusions: We observed that larger differences in methylation levels between individuals were often coupled to allele-specific methylation and genetic polymorphisms, suggesting that the inter-individual variability of DNA methylation is strongly influenced by genetic differences. Therefore, genetic differences must be taken into account in future comparative DNA methylation studies.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
