

Proteasome inhibition suppresses DNA-dependent protein kinase activation caused by camptothecin
- PMID: 19959400
- PMCID: PMC2818427
- DOI: 10.1016/j.dnarep.2009.10.008
Proteasome inhibition suppresses DNA-dependent protein kinase activation caused by camptothecin
Abstract
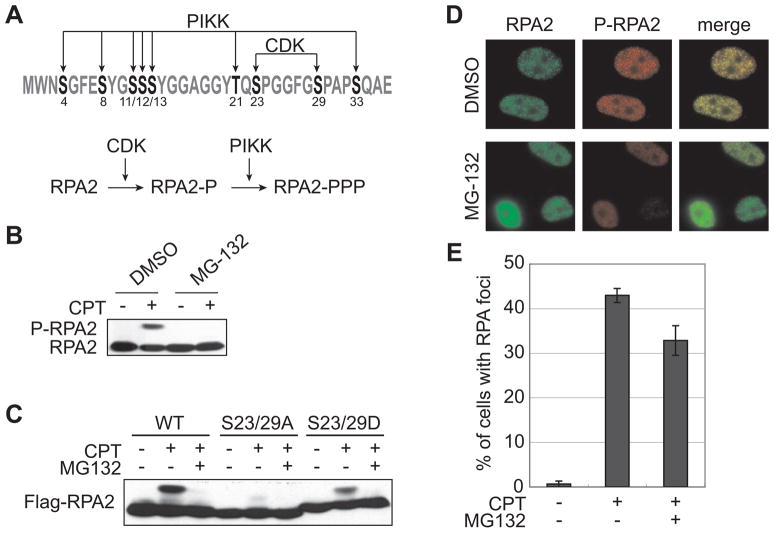
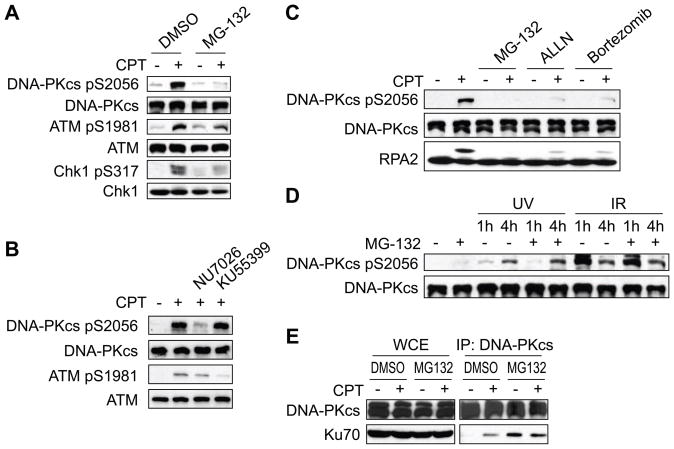
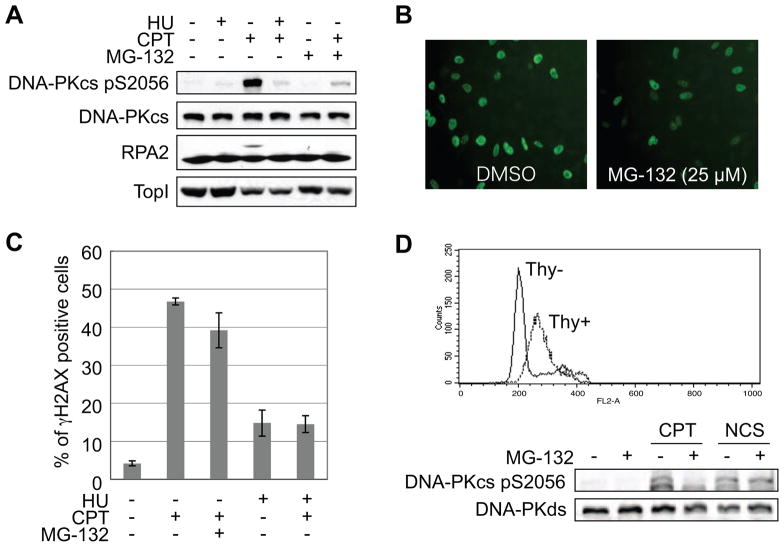
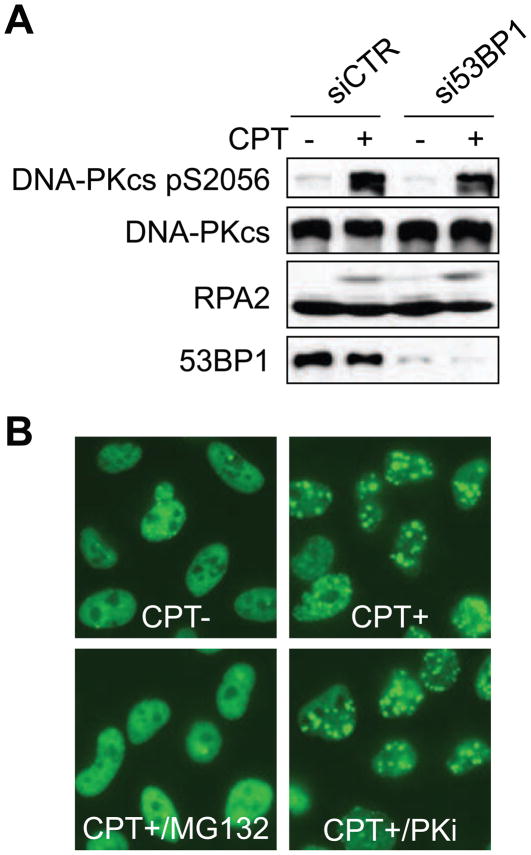
The ubiquitin-proteasome pathway plays an important role in DNA damage signaling and repair by facilitating the recruitment and activation of DNA repair factors and signaling proteins at sites of damaged chromatin. Proteasome activity is generally not thought to be required for activation of apical signaling kinases including the PI3K-related kinases (PIKKs) ATM, ATR, and DNA-PK that orchestrate downstream signaling cascades in response to diverse genotoxic stimuli. In a previous work, we showed that inhibition of the proteasome by MG-132 suppressed 53BP1 (p53 binding protein1) phosphorylation as well as RPA2 (replication protein A2) phosphorylation in response to the topoisomerase I (TopI) poison camptothecin (CPT). To address the mechanism of proteasome-dependent RPA2 phosphorylation, we investigated the effects of proteasome inhibitors on the upstream PIKKs. MG-132 sharply suppressed CPT-induced DNA-PKcs autophosphorylation, a marker of the activation, whereas the phosphorylation of ATM and ATR substrates was only slightly suppressed by MG-132, suggesting that DNA-PK among the PIKKs is specifically regulated by the proteasome in response to CPT. On the other hand, MG-132 did not suppress DNA-PK activation in response to UV or IR. MG-132 blocked the interaction between DNA-PKcs and Ku heterodimer enhanced by CPT, and hydroxyurea pre-treatment completely abolished CPT-induced DNA-PKcs autophosphorylation, indicating a requirement for ongoing DNA replication. CPT-induced TopI degradation occurred independent of DNA-PK activation, suggesting that DNA-PK activation does not require degradation of trapped TopI complexes. The combined results suggest that CPT-dependent replication fork collapse activates DNA-PK signaling through a proteasome dependent, TopI degradation-independent pathway. The implications of DNA-PK activation in the context of TopI poison-based therapies are discussed.
Copyright (c) 2009 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
Similar articles
-
Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison.J Biol Chem. 2010 May 14;285(20):15201-15208. doi: 10.1074/jbc.M110.101808. Epub 2010 Mar 19. J Biol Chem. 2010. PMID: 20304914 Free PMC article.
-
Differential involvement of phosphatidylinositol 3-kinase-related protein kinases in hyperphosphorylation of replication protein A2 in response to replication-mediated DNA double-strand breaks.Genes Cells. 2006 Mar;11(3):237-46. doi: 10.1111/j.1365-2443.2006.00942.x. Genes Cells. 2006. PMID: 16483312
-
Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes.EMBO J. 1999 Mar 1;18(5):1397-406. doi: 10.1093/emboj/18.5.1397. EMBO J. 1999. PMID: 10064605 Free PMC article.
-
A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation.DNA Repair (Amst). 2010 Dec 10;9(12):1307-14. doi: 10.1016/j.dnarep.2010.09.019. Epub 2010 Oct 28. DNA Repair (Amst). 2010. PMID: 21030321 Free PMC article. Review.
-
ATM, ATR and DNA-PKcs kinases-the lessons from the mouse models: inhibition ≠ deletion.Cell Biosci. 2020 Jan 29;10:8. doi: 10.1186/s13578-020-0376-x. eCollection 2020. Cell Biosci. 2020. PMID: 32015826 Free PMC article. Review.
Cited by
-
Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison.J Biol Chem. 2010 May 14;285(20):15201-15208. doi: 10.1074/jbc.M110.101808. Epub 2010 Mar 19. J Biol Chem. 2010. PMID: 20304914 Free PMC article.
-
APOBEC3B reporter myeloma cell lines identify DNA damage response pathways leading to APOBEC3B expression.PLoS One. 2020 Jan 8;15(1):e0223463. doi: 10.1371/journal.pone.0223463. eCollection 2020. PLoS One. 2020. PMID: 31914134 Free PMC article.
-
DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair.Oncogene. 2013 May 9;32(19):2452-62. doi: 10.1038/onc.2012.257. Epub 2012 Jul 16. Oncogene. 2013. PMID: 22797063 Free PMC article.
-
Proteasome inhibition induces DNA damage and reorganizes nuclear architecture and protein synthesis machinery in sensory ganglion neurons.Cell Mol Life Sci. 2014 May;71(10):1961-75. doi: 10.1007/s00018-013-1474-2. Epub 2013 Sep 24. Cell Mol Life Sci. 2014. PMID: 24061536 Free PMC article.
-
Rapid Diminution in the Level and Activity of DNA-Dependent Protein Kinase in Cancer Cells by a Reactive Nitro-Benzoxadiazole Compound.Int J Mol Sci. 2016 May 11;17(5):703. doi: 10.3390/ijms17050703. Int J Mol Sci. 2016. PMID: 27187356 Free PMC article.
References
-
- Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell. 2004;118:9–17. - PubMed
-
- Binz SK, Sheehan AM, Wold MS. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair. 2004;3:1015–1024. - PubMed
-
- Wang Y, Perrault AR, Iliakis G. Down-regulation of DNA replication in extracts of camptothecin-treated cells: activation of an S-phase checkpoint? Cancer Res. 1997;57:1654–1659. - PubMed
-
- Sakasai R, Shinohe K, Ichijima Y, Okita N, Shibata A, Asahina K, Teraoka H. Differential involvement of phosphatidylinositol 3-kinase-related protein kinases in hyperphosphorylation of replication protein A2 in response to replication-mediated DNA double-strand breaks. Genes Cells. 2006;11:237–246. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
