

Gap junctions sensitize cancer cells to proteasome inhibitor MG132-induced apoptosis
- PMID: 19961488
- PMCID: PMC11159102
- DOI: 10.1111/j.1349-7006.2009.01421.x
Gap junctions sensitize cancer cells to proteasome inhibitor MG132-induced apoptosis
Abstract
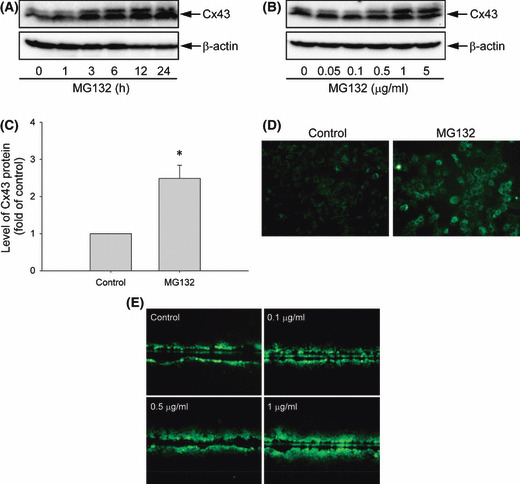
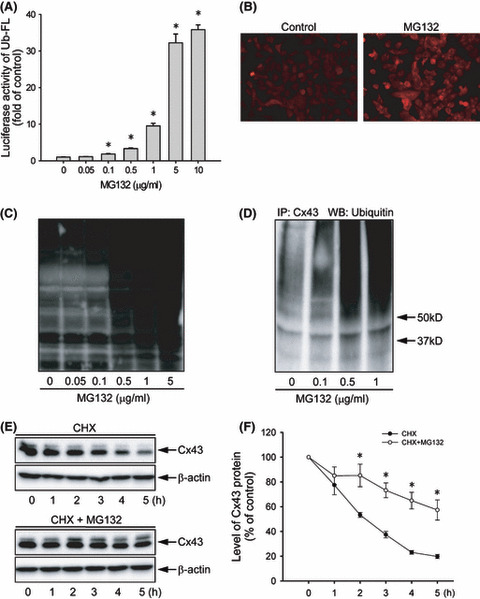
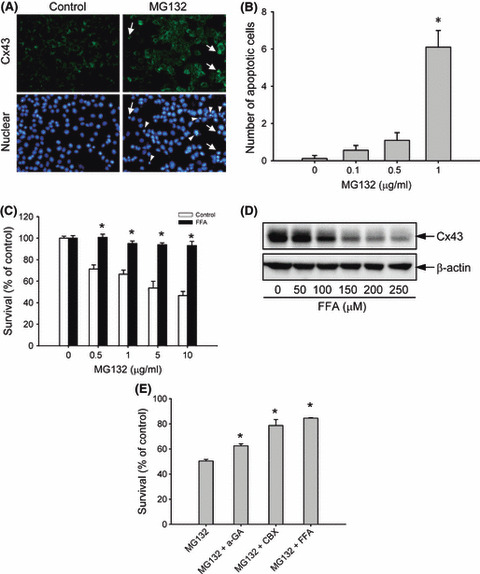
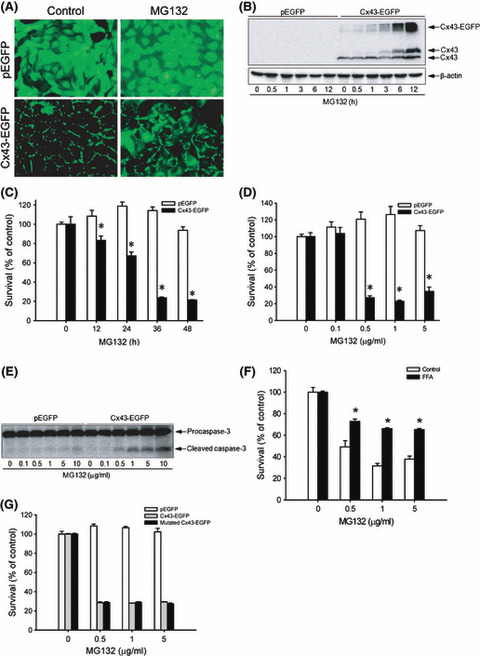
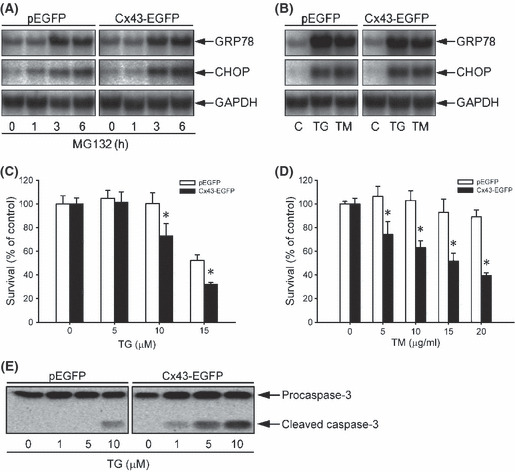
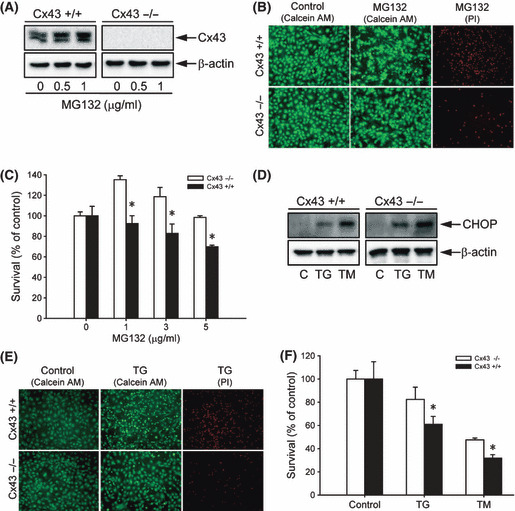
Proteasome inhibition is a promising approach for cancer therapy. However, the mechanisms involved have not been fully elucidated. Gap junctions play important roles in the regulation of tumor cell phenotypes and mediation of the bystander effect in cancer therapy. Because the degradation of gap junction proteins involves the proteasome, we speculated that altered gap junctions might contribute to the antitumor activities of proteasome inhibition. Incubation of Hepa-1c1c7 cells with the proteasome inhibitor MG132 elevated the levels of gap junction protein connexin 43 (Cx43) and promoted gap junctional intercellular communication. This was associated with a marked accumulation of ubiquitylated Cx43 and a significantly decreased rate of Cx43 degradation. The elevated Cx43 contributed to MG132-induced cell apoptosis. This is shown by the observations that: (i) overexpression of Cx43 in the gap junction-deficient LLC-PK1 cells rendered them vulnerable to MG132-elicited cell injury; (ii) fibroblasts derived from Cx43-null mice were more resistant to MG-132 compared with Cx43 wild-type control; and (iii) the gap junction inhibitor flufenamic acid significantly attenuated cell damage caused by MG132 in Hepa-1c1c7 cells. Further studies demonstrated that MG132 activates endoplasmic reticulum stress. Exposure of cells to the endoplasmic reticulum stress inducers thapsigargin and tunicamycin also led to cell apoptosis, which was modulated by Cx43 levels in a way similar to MG132. These results suggested that elevated Cx43 sensitizes cells to MG132-induced cell apoptosis. Regulation of gap junctions could be an important mechanism behind the antitumor activities of proteasome inhibitors.
Figures
Similar articles
-
Role of the ubiquitin-proteasome pathway in downregulation of the gap-junction protein Connexin43 by TNF-{alpha} in human corneal fibroblasts.Invest Ophthalmol Vis Sci. 2010 Apr;51(4):1943-7. doi: 10.1167/iovs.09-3573. Epub 2009 Nov 11. Invest Ophthalmol Vis Sci. 2010. PMID: 19907029
-
Disruption of gap junctions attenuates aminoglycoside-elicited renal tubular cell injury.Br J Pharmacol. 2010 Aug;160(8):2055-68. doi: 10.1111/j.1476-5381.2010.00860.x. Br J Pharmacol. 2010. PMID: 20649601 Free PMC article.
-
Phosphorylation of connexin 43 acts as a stimuli for proteasome-dependent degradation of the protein in lens epithelial cells.Mol Vis. 2003 Jan 30;9:24-30. Mol Vis. 2003. PMID: 12567182
-
[Remodeling of cardiac gap junctions and arrhythmias].Sheng Li Xue Bao. 2011 Dec 25;63(6):586-92. Sheng Li Xue Bao. 2011. PMID: 22193455 Review. Chinese.
-
Connexin 43 (Cx43) in cancer: Implications for therapeutic approaches via gap junctions.Cancer Lett. 2019 Feb 1;442:439-444. doi: 10.1016/j.canlet.2018.10.043. Epub 2018 Nov 22. Cancer Lett. 2019. PMID: 30472182 Review.
Cited by
-
Ubiquitination, intracellular trafficking, and degradation of connexins.Arch Biochem Biophys. 2012 Aug 1;524(1):16-22. doi: 10.1016/j.abb.2011.12.027. Epub 2012 Jan 3. Arch Biochem Biophys. 2012. PMID: 22239989 Free PMC article. Review.
-
Matrine inhibits prostate cancer via activation of the unfolded protein response/endoplasmic reticulum stress signaling and reversal of epithelial to mesenchymal transition.Mol Med Rep. 2018 Jul;18(1):945-957. doi: 10.3892/mmr.2018.9061. Epub 2018 May 23. Mol Med Rep. 2018. PMID: 29845238 Free PMC article.
-
MicroRNA-30/Cx43 axis contributes to podocyte injury by regulating ER stress in diabetic nephropathy.Ann Transl Med. 2020 Dec;8(24):1674. doi: 10.21037/atm-20-6989. Ann Transl Med. 2020. PMID: 33490186 Free PMC article.
-
Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability.J Biol Chem. 2012 Jan 20;287(4):2600-7. doi: 10.1074/jbc.M111.276261. Epub 2011 Dec 2. J Biol Chem. 2012. PMID: 22139843 Free PMC article.
-
Degradation of connexins through the proteasomal, endolysosomal and phagolysosomal pathways.J Membr Biol. 2012 Jul;245(7):389-400. doi: 10.1007/s00232-012-9461-3. Epub 2012 Jul 8. J Membr Biol. 2012. PMID: 22772442 Review.
References
-
- Daviet L, Colland F. Targeting ubiquitin specific proteases for drug discovery. Biochimie 2008; 90: 270–83. - PubMed
-
- Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004; 5: 417–21. - PubMed
-
- Hideshima T, Richardson P, Chauhan D et al. The proteasome inhibitor PS‐341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001; 61: 3071–6. - PubMed
-
- Conticello C, Adamo L, Giuffrida R et al. Proteasome inhibitors synergize with tumor necrosis factor‐related apoptosis‐induced ligand to induce anaplastic thyroid carcinoma cell death. J Clin Endocrinol Metab 2007; 92: 1938–42. - PubMed
-
- Cusack JC Jr, Liu R, Houston M et al. Enhanced chemosensitivity to CPT‐11 with proteasome inhibitor PS‐341: implications for systemic nuclear factor‐kappaB inhibition. Cancer Res 2001; 61: 3535–40. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
