

Multiple non-cell-autonomous defects underlie neocortical callosal dysgenesis in Nfib-deficient mice
- PMID: 19961580
- PMCID: PMC2802587
- DOI: 10.1186/1749-8104-4-43
Multiple non-cell-autonomous defects underlie neocortical callosal dysgenesis in Nfib-deficient mice
Abstract
Background: Agenesis of the corpus callosum is associated with many human developmental syndromes. Key mechanisms regulating callosal formation include the guidance of axons arising from pioneering neurons in the cingulate cortex and the development of cortical midline glial populations, but their molecular regulation remains poorly characterised. Recent data have shown that mice lacking the transcription factor Nfib exhibit callosal agenesis, yet neocortical callosal neurons express only low levels of Nfib. Therefore, we investigate here how Nfib functions to regulate non-cell-autonomous mechanisms of callosal formation.
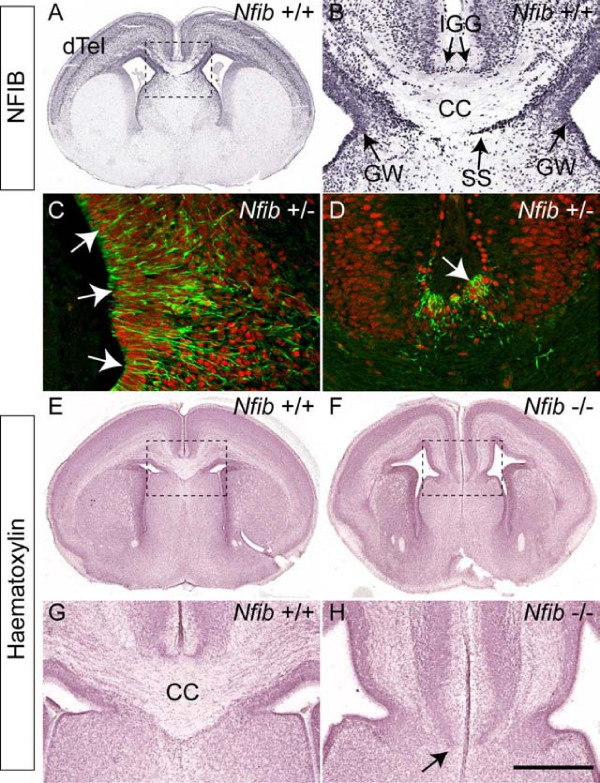
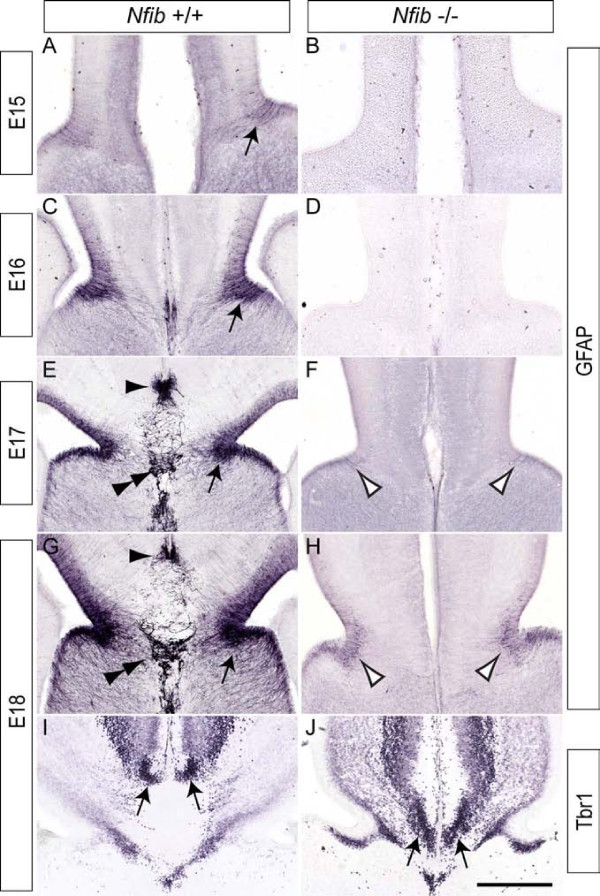
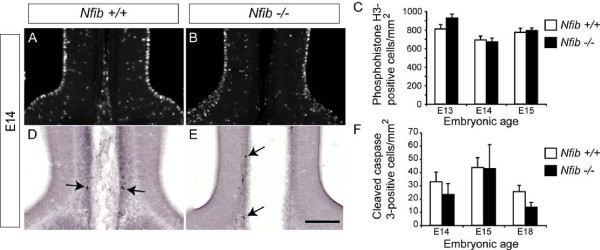
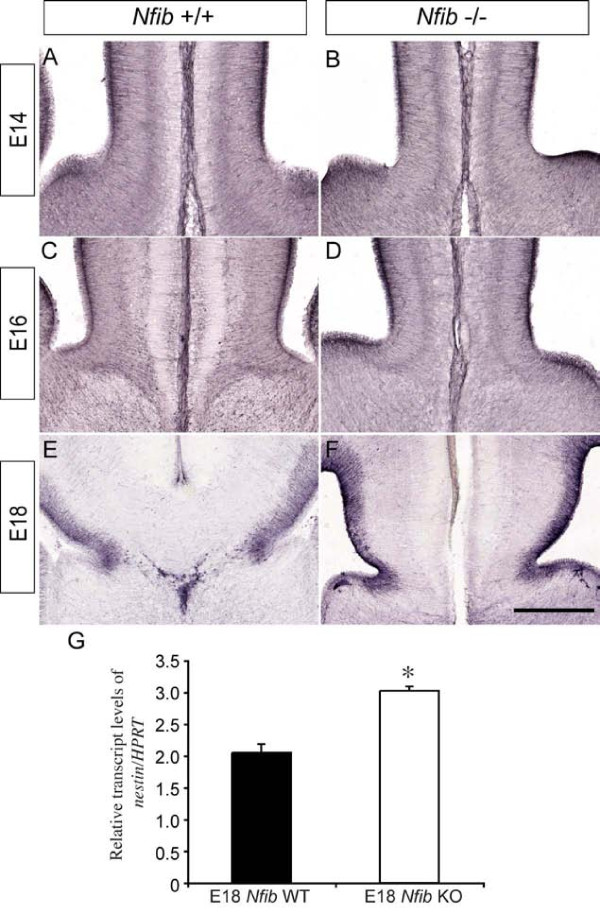
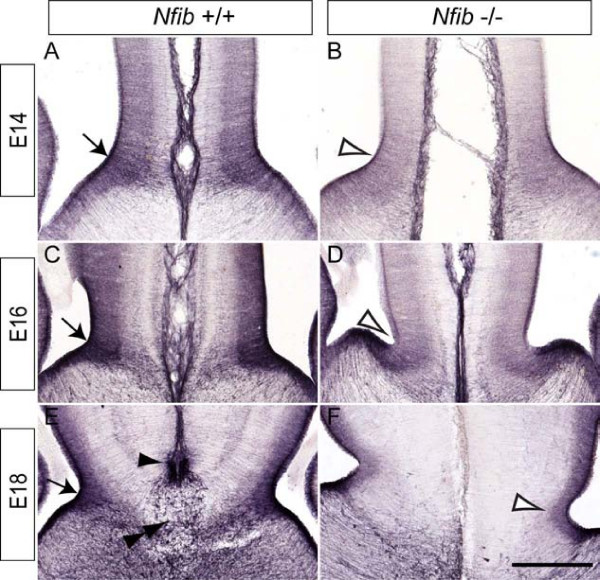
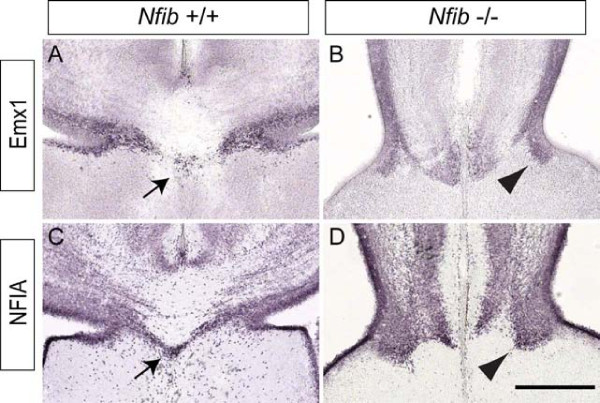
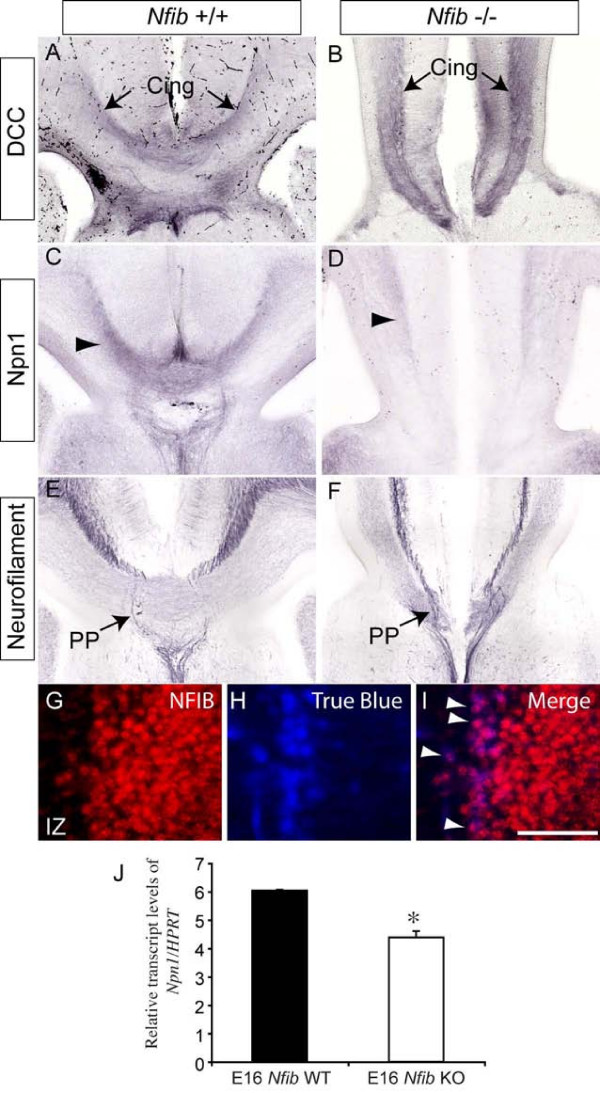
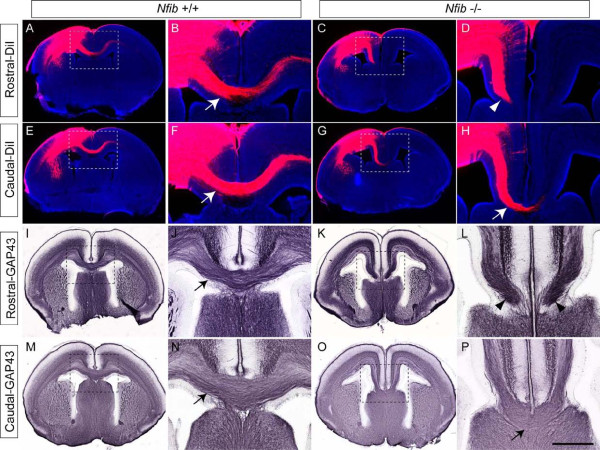
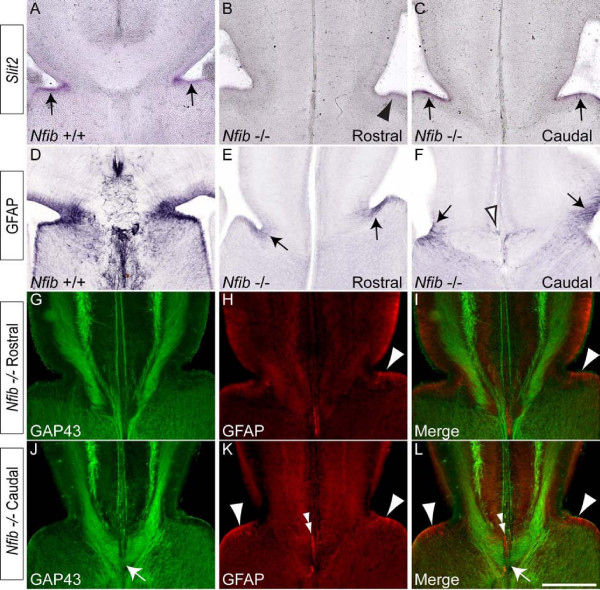
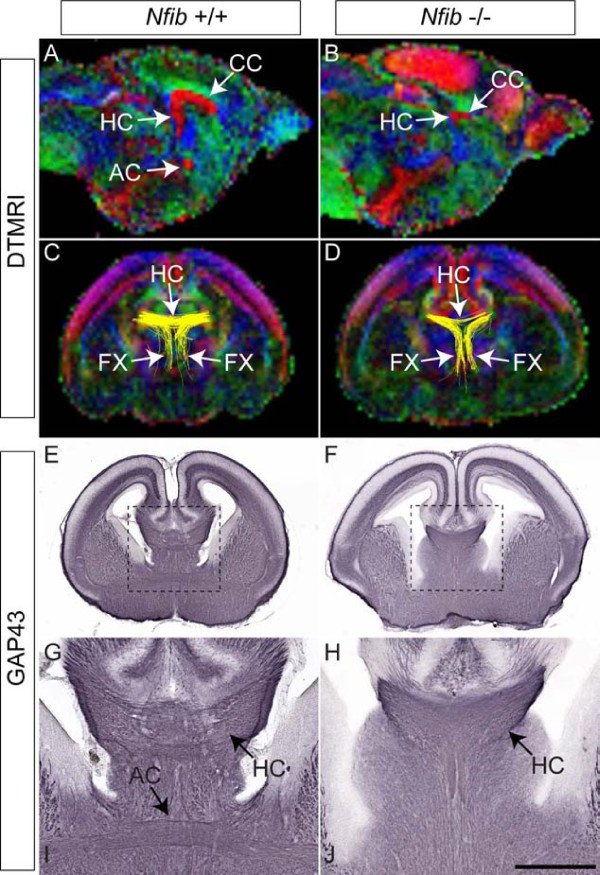
Results: Our investigations confirmed a reduction in glial cells at the midline in Nfib-/- mice. To determine how this occurs, we examined radial progenitors at the cortical midline and found that they were specified correctly in Nfib mutant mice, but did not differentiate into mature glia. Cellular proliferation and apoptosis occurred normally at the midline of Nfib mutant mice, indicating that the decrease in midline glia observed was due to deficits in differentiation rather than proliferation or apoptosis. Next we investigated the development of callosal pioneering axons in Nfib-/- mice. Using retrograde tracer labelling, we found that Nfib is expressed in cingulate neurons and hence may regulate their development. In Nfib-/- mice, neuropilin 1-positive axons fail to cross the midline and expression of neuropilin 1 is diminished. Tract tracing and immunohistochemistry further revealed that, in late gestation, a minor population of neocortical axons does cross the midline in Nfib mutants on a C57Bl/6J background, forming a rudimentary corpus callosum. Finally, the development of other forebrain commissures in Nfib-deficient mice is also aberrant.
Conclusion: The formation of the corpus callosum is severely delayed in the absence of Nfib, despite Nfib not being highly expressed in neocortical callosal neurons. Our results indicate that in addition to regulating the development of midline glial populations, Nfib also regulates the expression of neuropilin 1 within the cingulate cortex. Collectively, these data indicate that defects in midline glia and cingulate cortex neurons are associated with the callosal dysgenesis seen in Nfib-deficient mice, and provide insight into how the development of these cellular populations is controlled at a molecular level.
Figures
Similar articles
-
A role for cingulate pioneering axons in the development of the corpus callosum.J Comp Neurol. 2001 May 28;434(2):147-57. doi: 10.1002/cne.1170. J Comp Neurol. 2001. PMID: 11331522
-
Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice.J Neurosci. 2003 Jan 1;23(1):203-12. doi: 10.1523/JNEUROSCI.23-01-00203.2003. J Neurosci. 2003. PMID: 12514217 Free PMC article.
-
PlexinA1 is crucial for the midline crossing of callosal axons during corpus callosum development in BALB/cAJ mice.PLoS One. 2019 Aug 20;14(8):e0221440. doi: 10.1371/journal.pone.0221440. eCollection 2019. PLoS One. 2019. PMID: 31430342 Free PMC article.
-
Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human.Clin Genet. 2004 Oct;66(4):276-89. doi: 10.1111/j.1399-0004.2004.00354.x. Clin Genet. 2004. PMID: 15355427 Review.
-
Axon guidance mechanisms for establishment of callosal connections.Neural Plast. 2013;2013:149060. doi: 10.1155/2013/149060. Epub 2013 Feb 24. Neural Plast. 2013. PMID: 23533817 Free PMC article. Review.
Cited by
-
NFIB Haploinsufficiency Is Associated with Intellectual Disability and Macrocephaly.Am J Hum Genet. 2018 Nov 1;103(5):752-768. doi: 10.1016/j.ajhg.2018.10.006. Am J Hum Genet. 2018. PMID: 30388402 Free PMC article.
-
A cascade of morphogenic signaling initiated by the meninges controls corpus callosum formation.Neuron. 2012 Feb 23;73(4):698-712. doi: 10.1016/j.neuron.2011.11.036. Neuron. 2012. PMID: 22365545 Free PMC article.
-
Combined allelic dosage of Nfia and Nfib regulates cortical development.Brain Neurosci Adv. 2017 Nov 22;1:2398212817739433. doi: 10.1177/2398212817739433. eCollection 2017 Jan-Dec. Brain Neurosci Adv. 2017. PMID: 32166136 Free PMC article.
-
NFIA controls telencephalic progenitor cell differentiation through repression of the Notch effector Hes1.J Neurosci. 2010 Jul 7;30(27):9127-39. doi: 10.1523/JNEUROSCI.6167-09.2010. J Neurosci. 2010. PMID: 20610746 Free PMC article.
-
19p13 microduplications encompassing NFIX are responsible for intellectual disability, short stature and small head circumference.Eur J Hum Genet. 2018 Jan;26(1):85-93. doi: 10.1038/s41431-017-0037-7. Epub 2017 Nov 28. Eur J Hum Genet. 2018. PMID: 29184170 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
