

Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies
- PMID: 19961595
- PMCID: PMC2797771
- DOI: 10.1186/1742-4690-6-111
Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies
Abstract
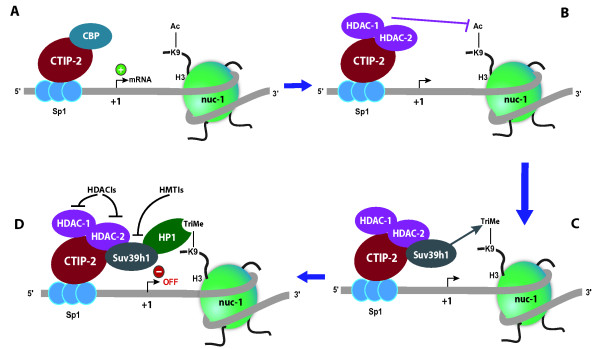
The persistence of HIV-1 latent reservoirs represents a major barrier to virus eradication in infected patients under HAART since interruption of the treatment inevitably leads to a rebound of plasma viremia. Latency establishes early after infection notably (but not only) in resting memory CD4+ T cells and involves numerous host and viral trans-acting proteins, as well as processes such as transcriptional interference, RNA silencing, epigenetic modifications and chromatin organization. In order to eliminate latent reservoirs, new strategies are envisaged and consist of reactivating HIV-1 transcription in latently-infected cells, while maintaining HAART in order to prevent de novo infection. The difficulty lies in the fact that a single residual latently-infected cell can in theory rekindle the infection. Here, we review our current understanding of the molecular mechanisms involved in the establishment and maintenance of HIV-1 latency and in the transcriptional reactivation from latency. We highlight the potential of new therapeutic strategies based on this understanding of latency. Combinations of various compounds used simultaneously allow for the targeting of transcriptional repression at multiple levels and can facilitate the escape from latency and the clearance of viral reservoirs. We describe the current advantages and limitations of immune T-cell activators, inducers of the NF-kappaB signaling pathway, and inhibitors of deacetylases and histone- and DNA- methyltransferases, used alone or in combinations. While a solution will not be achieved by tomorrow, the battle against HIV-1 latent reservoirs is well- underway.
Figures
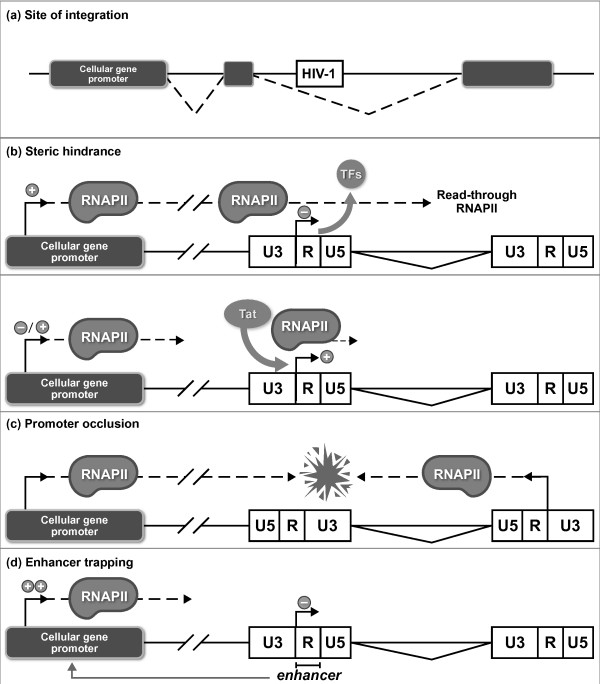
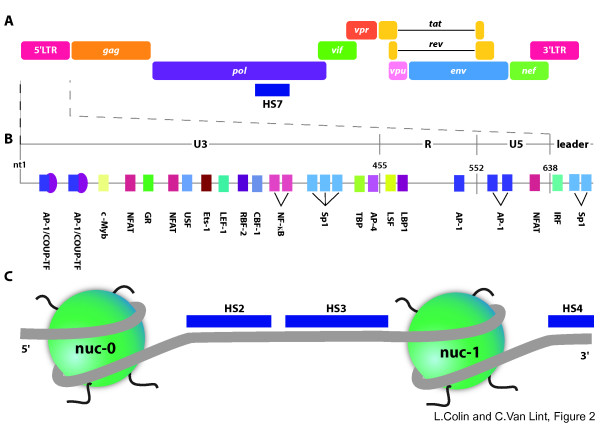
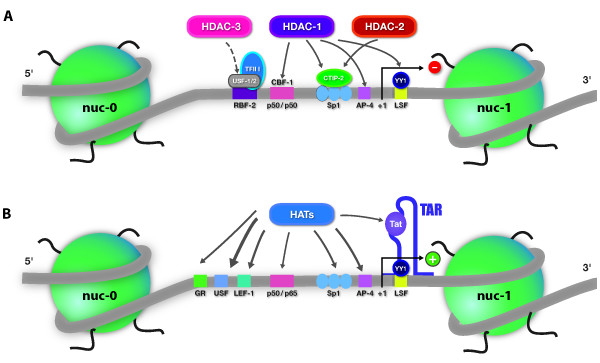
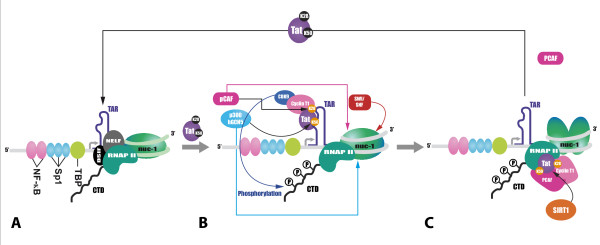
References
-
- Nijhuis M, van Maarseveen NM, Boucher CA. Antiviral resistance and impact on viral replication capacity: evolution of viruses under antiviral pressure occurs in three phases. Handb Exp Pharmacol. 2009. pp. 299–320. full_text. - PubMed
-
- Dahl V, Josefsson L, Palmer S. HIV reservoirs, latency, and reactivation: Prospects for eradication. Antiviral Res. 2009. in press . - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
