

Structural and biophysical studies of human PARP-1 in complex with damaged DNA
- PMID: 19962992
- PMCID: PMC2864582
- DOI: 10.1016/j.jmb.2009.11.062
Structural and biophysical studies of human PARP-1 in complex with damaged DNA
Abstract
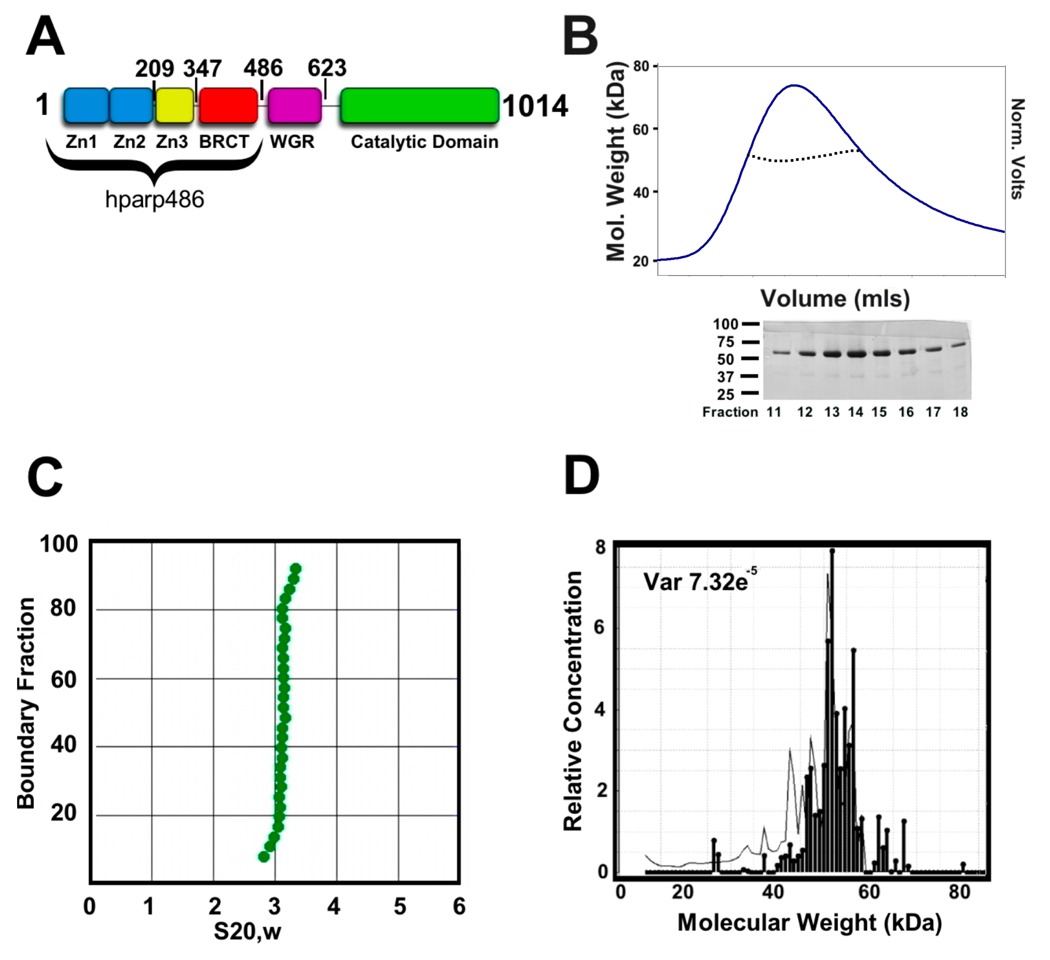
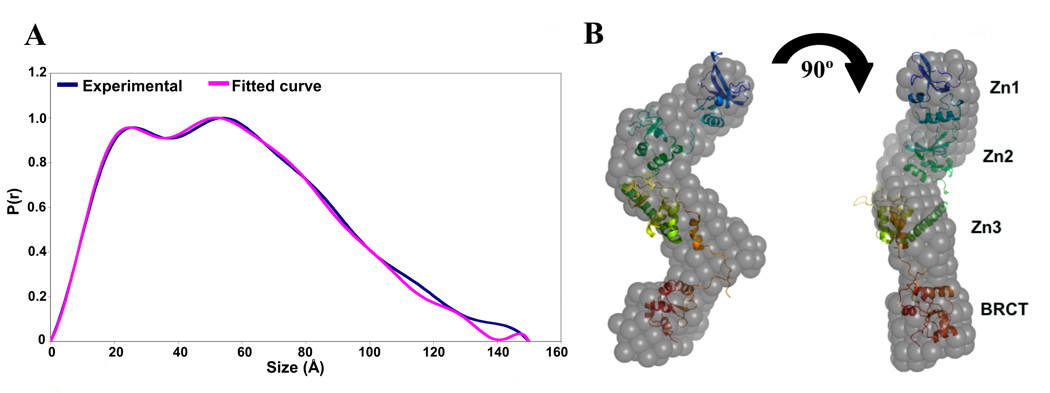
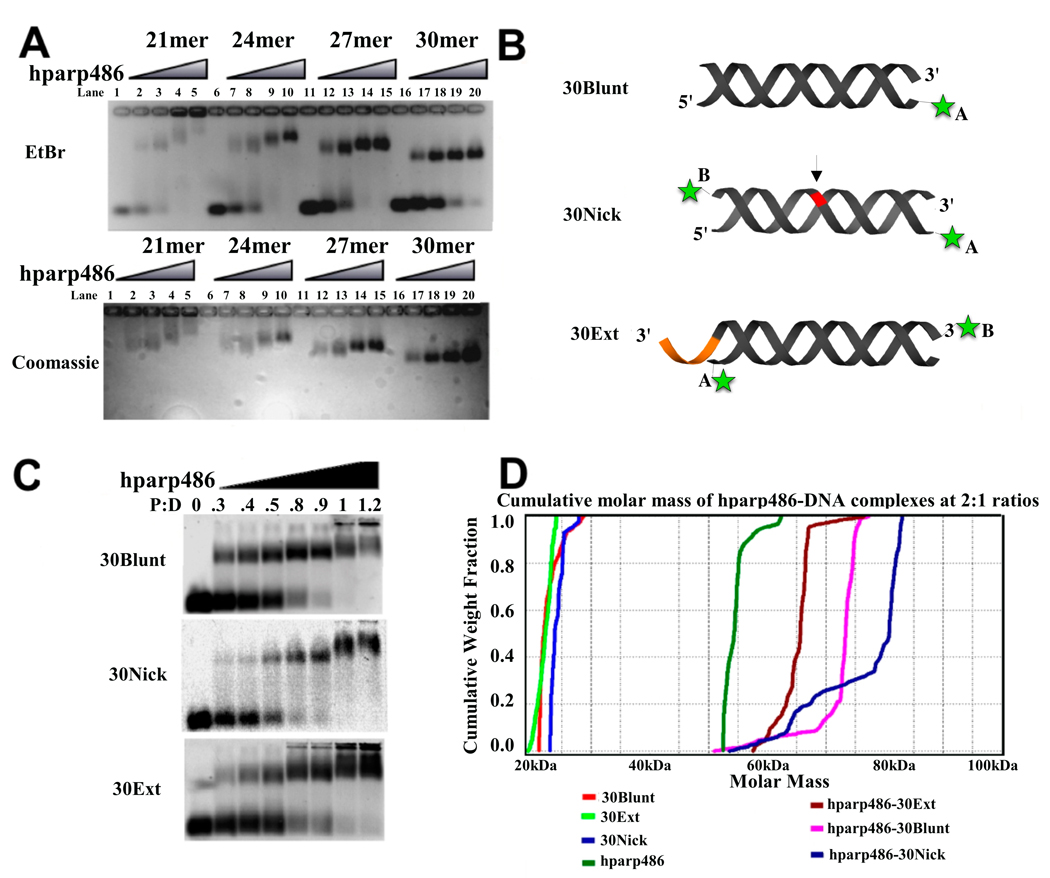
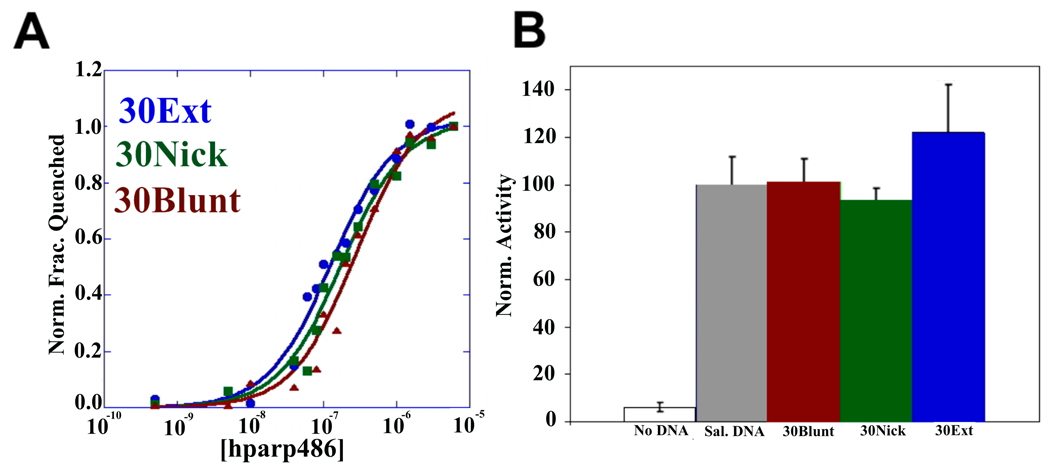
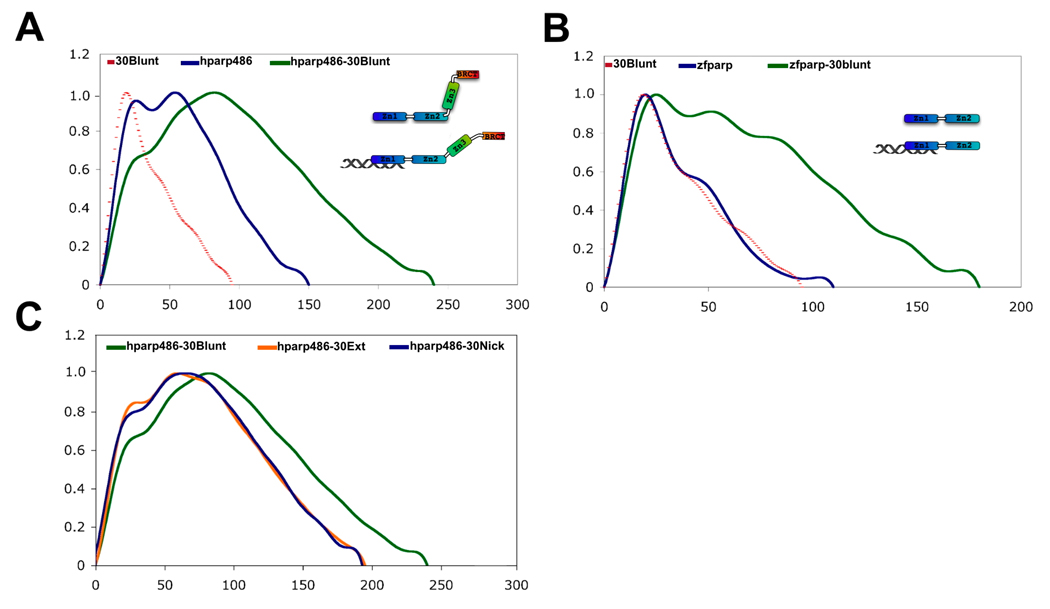
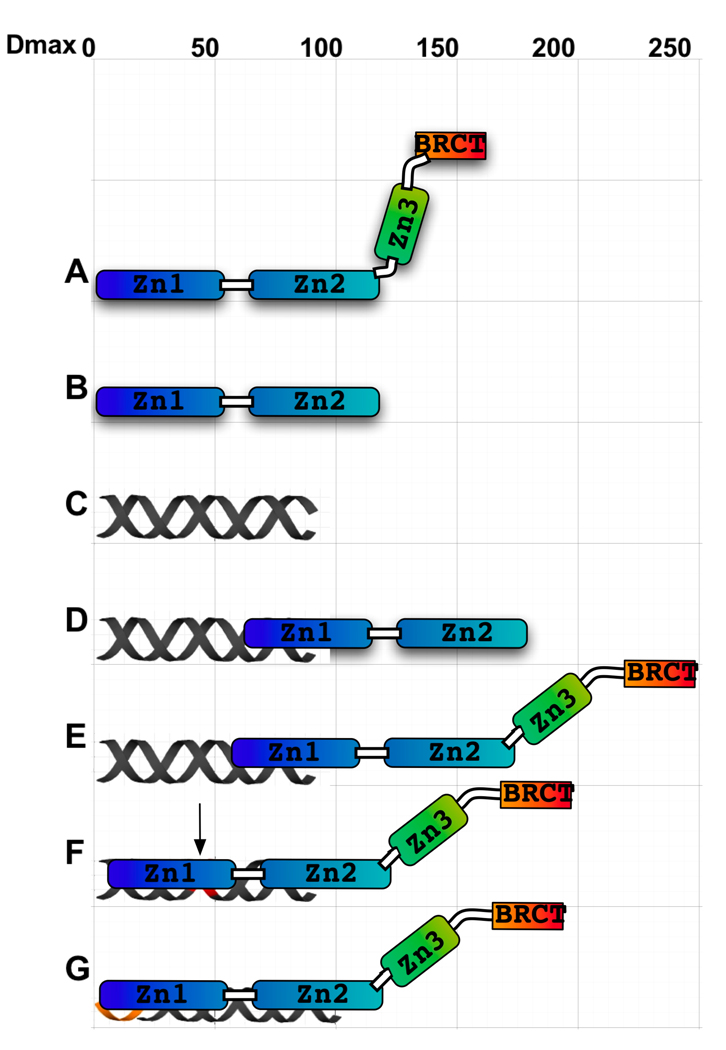
The enzyme poly(ADP-ribose) polymerase-1 (PARP-1) is a global monitor of chromatin structure and DNA damage repair. PARP-1 binds to nucleosomes and poly(ADP-ribosylates) histones and several chromatin-associated factors to expose specific DNA sequences to the cellular machinery involved in gene transcription and/or DNA damage repair. While these processes are critical to genomic stability, the molecular mechanisms of how DNA damage induces PARP-1 activation are poorly understood. We have used biochemical and thermodynamic measurements in conjunction with small-angle X-ray scattering to determine the stoichiometry, affinity, and overall structure of a human PARP-1 construct containing the entire DNA binding region, the zinc ribbon domain, and automodification domains (residues 1-486). The interaction of this PARP-1 protein construct with three different DNA damage models (DNA constructs containing a nick, a blunt end, or a 3' extension) was evaluated. Our data indicate that PARP-1 binds each DNA damage model as a monomer and with similar affinity, in all cases resulting in robust activation of the catalytic domain. Using small-angle X-ray scattering, we determined that the N-terminal half of PARP-1 behaves as an extended and flexible arrangement of individually folded domains in the absence of DNA. Upon binding DNA, PARP-1 undergoes a conformational change in the area surrounding the zinc ribbon domain. These data support a model in which PARP-1, upon binding DNA, undergoes a conformational change to become an active nuclear enzyme.
Copyright 2009 Elsevier Ltd. All rights reserved.
Figures
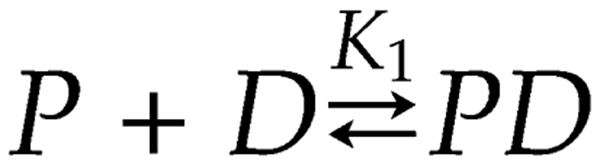
Similar articles
-
Poly(ADP-ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer.Cochrane Database Syst Rev. 2022 Feb 16;2(2):CD007929. doi: 10.1002/14651858.CD007929.pub4. Cochrane Database Syst Rev. 2022. PMID: 35170751 Free PMC article.
-
The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review.Int J Mol Sci. 2022 Jul 23;23(15):8125. doi: 10.3390/ijms23158125. Int J Mol Sci. 2022. PMID: 35897700 Free PMC article.
-
Crystal structure and mutagenesis of a nucleic acid-binding BRCT domain in human PARP4.J Biol Chem. 2025 Jun;301(6):110277. doi: 10.1016/j.jbc.2025.110277. Epub 2025 May 22. J Biol Chem. 2025. PMID: 40412520 Free PMC article.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Novel inhibitors of PARP1 and PARP14: design, synthesis, and potentiation of cisplatin efficacy in cancer.Future Med Chem. 2025 Jan;17(1):35-58. doi: 10.1080/17568919.2024.2437972. Epub 2024 Dec 18. Future Med Chem. 2025. PMID: 39691063
Cited by
-
Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins.Nucleic Acids Res. 2019 May 7;47(8):3811-3827. doi: 10.1093/nar/gkz120. Nucleic Acids Res. 2019. PMID: 30799503 Free PMC article. Review.
-
HPF1 Regulates Pol β Efficiency in Nucleosomes via the Modulation of Total Poly(ADP-Ribose) Synthesis.Int J Mol Sci. 2025 Feb 20;26(5):1794. doi: 10.3390/ijms26051794. Int J Mol Sci. 2025. PMID: 40076422 Free PMC article.
-
PARP inhibition: PARP1 and beyond.Nat Rev Cancer. 2010 Apr;10(4):293-301. doi: 10.1038/nrc2812. Epub 2010 Mar 4. Nat Rev Cancer. 2010. PMID: 20200537 Free PMC article. Review.
-
Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1.Mol Cell. 2015 Dec 3;60(5):742-754. doi: 10.1016/j.molcel.2015.10.032. Epub 2015 Nov 25. Mol Cell. 2015. PMID: 26626479 Free PMC article.
-
Dynamics of the HD regulatory subdomain of PARP-1; substrate access and allostery in PARP activation and inhibition.Nucleic Acids Res. 2021 Feb 26;49(4):2266-2288. doi: 10.1093/nar/gkab020. Nucleic Acids Res. 2021. PMID: 33511412 Free PMC article.
References
-
- Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. - PubMed
-
- Bouchard VJ, Rouleau M, Poirier GG. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol. 2003;31:446–454. - PubMed
-
- Jacobson EL, Antol KM, Juarez-Salinas H, Jacobson MK. Poly(ADP-ribose) metabolism in ultraviolet irradiated human fibroblasts. J Biol Chem. 1983;258:103–107. - PubMed
-
- Oleinick NL, Evans HH. Poly(ADP-ribose) and the response of cells to ionizing radiation. Radiat Res. 1985;101:29–46. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
