

Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug
- PMID: 19965621
- PMCID: PMC2844013
- DOI: 10.1182/blood-2009-04-213520
Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug
Abstract
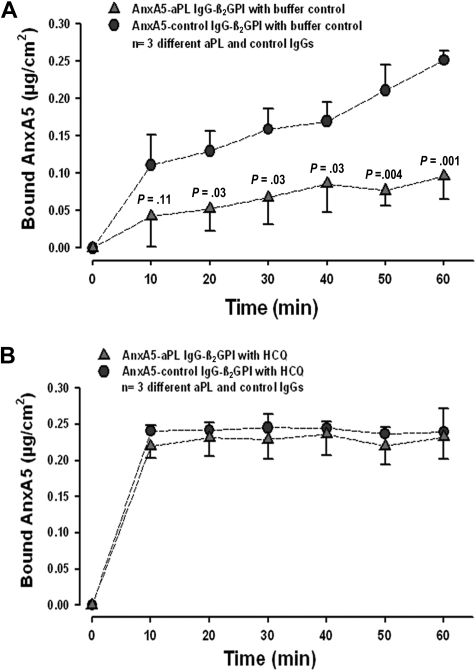
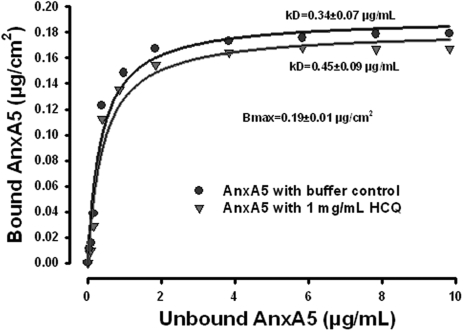
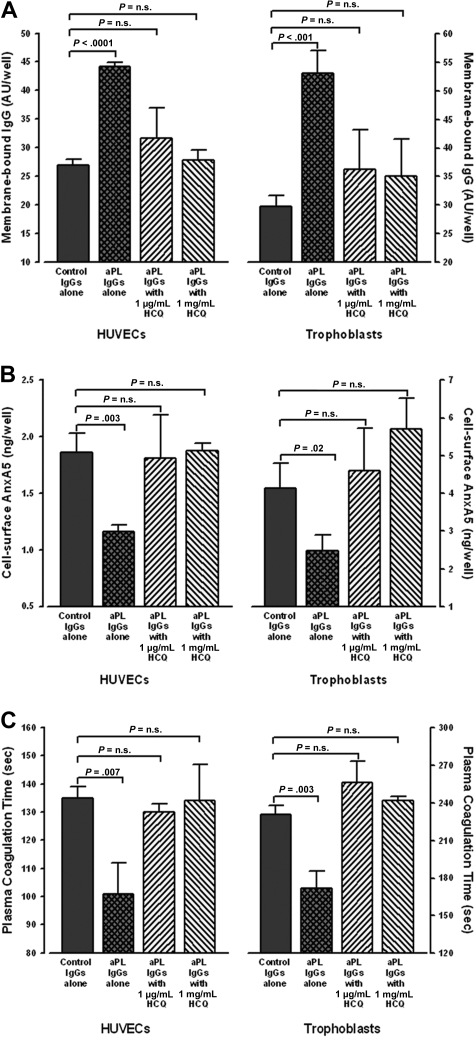
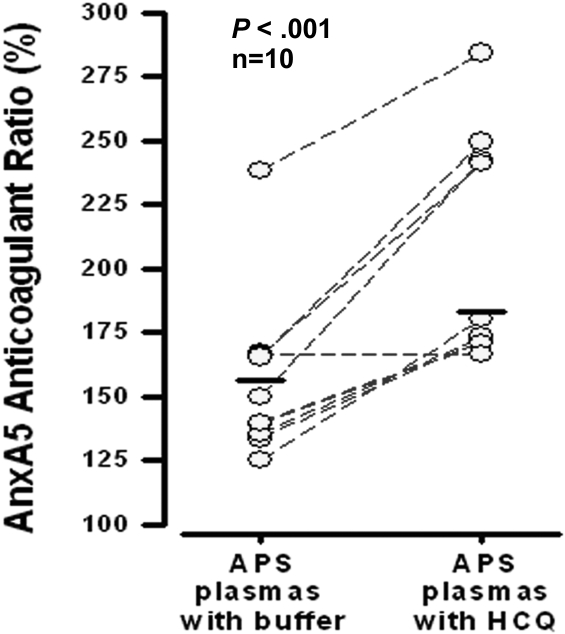
Annexin A5 (AnxA5) is a potent anticoagulant protein that crystallizes over phospholipid bilayers (PLBs), blocking their availability for coagulation reactions. Antiphospholipid antibodies disrupt AnxA5 binding, thereby accelerating coagulation reactions. This disruption may contribute to thrombosis and miscarriages in the antiphospholipid syndrome (APS). We investigated whether the antimalarial drug, hydroxychloroquine (HCQ), might affect this prothrombotic mechanism. Binding of AnxA5 to PLBs was measured with labeled AnxA5 and also imaged with atomic force microscopy. Immunoglobulin G levels, AnxA5, and plasma coagulation times were measured on cultured human umbilical vein endothelial cells and a syncytialized trophoblast cell line. AnxA5 anticoagulant activities of APS patient plasmas were also determined. HCQ reversed the effect of antiphospholipid antibodies on AnxA5 and restored AnxA5 binding to PLBs, an effect corroborated by atomic force microscopy. Similar reversals of antiphospholipid-induced abnormalities were measured on the surfaces of human umbilical vein endothelial cells and syncytialized trophoblast cell lines, wherein HCQ reduced the binding of antiphospholipid antibodies, increased cell-surface AnxA5 concentrations, and prolonged plasma coagulation to control levels. In addition, HCQ increased the AnxA5 anticoagulant activities of APS patient plasmas. In conclusion, HCQ reversed antiphospholipid-mediated disruptions of AnxA5 on PLBs and cultured cells, and in APS patient plasmas. These results support the concept of novel therapeutic approaches that address specific APS disease mechanisms.
Figures



Comment in
-
A novel antiphospholipid antibody agent?Blood. 2010 Mar 18;115(11):2124-5. doi: 10.1182/blood-2009-12-258889. Blood. 2010. PMID: 20299521 No abstract available.
Similar articles
-
The annexin A5-mediated pathogenic mechanism in the antiphospholipid syndrome: role in pregnancy losses and thrombosis.Lupus. 2010 Apr;19(4):460-9. doi: 10.1177/0961203310361485. Lupus. 2010. PMID: 20353989 Review.
-
Hydroxychloroquine directly reduces the binding of antiphospholipid antibody-beta2-glycoprotein I complexes to phospholipid bilayers.Blood. 2008 Sep 1;112(5):1687-95. doi: 10.1182/blood-2008-03-144204. Epub 2008 Jun 24. Blood. 2008. PMID: 18577708 Free PMC article.
-
Hydroxychloroquine reduces binding of antiphospholipid antibodies to syncytiotrophoblasts and restores annexin A5 expression.Am J Obstet Gynecol. 2011 Dec;205(6):576.e7-14. doi: 10.1016/j.ajog.2011.06.064. Epub 2011 Jun 24. Am J Obstet Gynecol. 2011. PMID: 21871597 Free PMC article.
-
Human monoclonal antiphospholipid antibodies disrupt the annexin A5 anticoagulant crystal shield on phospholipid bilayers: evidence from atomic force microscopy and functional assay.Am J Pathol. 2003 Sep;163(3):1193-200. doi: 10.1016/S0002-9440(10)63479-7. Am J Pathol. 2003. PMID: 12937161 Free PMC article.
-
Resistance to annexin A5 anticoagulant activity: a thrombogenic mechanism for the antiphospholipid syndrome.Lupus. 2008 Oct;17(10):922-30. doi: 10.1177/0961203308095029. Lupus. 2008. PMID: 18827057 Review.
Cited by
-
Association of Higher Hydroxychloroquine Blood Levels With Reduced Thrombosis Risk in Systemic Lupus Erythematosus.Arthritis Rheumatol. 2021 Jun;73(6):997-1004. doi: 10.1002/art.41621. Epub 2021 May 2. Arthritis Rheumatol. 2021. PMID: 33403833 Free PMC article.
-
Immunosuppressive Treatment in Antiphospholipid Syndrome: Is It Worth It?Biomedicines. 2021 Feb 1;9(2):132. doi: 10.3390/biomedicines9020132. Biomedicines. 2021. PMID: 33535377 Free PMC article. Review.
-
Differential inflammatory conditioning of the bone marrow by acute myeloid leukemia and its impact on progression.Blood Adv. 2024 Oct 8;8(19):4983-4996. doi: 10.1182/bloodadvances.2024012867. Blood Adv. 2024. PMID: 38996202 Free PMC article.
-
Resistance to annexin A5 anticoagulant activity in women with histories for obstetric antiphospholipid syndrome.Am J Obstet Gynecol. 2011 Nov;205(5):485.e17-23. doi: 10.1016/j.ajog.2011.06.019. Epub 2011 Jun 15. Am J Obstet Gynecol. 2011. PMID: 21784397 Free PMC article.
-
Hydroxychloroquine as an Immunomodulatory and Antithrombotic Treatment in Antiphospholipid Syndrome.Int J Mol Sci. 2023 Jan 10;24(2):1331. doi: 10.3390/ijms24021331. Int J Mol Sci. 2023. PMID: 36674847 Free PMC article. Review.
References
-
- Ginsberg JS, Wells PS, Brill Edwards P, et al. Antiphospholipid antibodies and venous thromboembolism. Blood. 1995;86:3685–3691. - PubMed
-
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. J Am Med Assoc. 2006;295:1050–1057. - PubMed
-
- Rand JH, Wu XX, Quinn AS, Taatjes DJ. Resistance to annexin A5 anticoagulant activity: a thrombogenic mechanism for the antiphospholipid syndrome. Lupus. 2008;17:922–930. - PubMed
-
- Andree HAM, Stuart MC, Hermens WT, et al. Clustering of lipid-bound annexin V may explain its anticoagulant effect. J Biol Chem. 1992;267:17907–17912. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
