

Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery
- PMID: 19965627
- PMCID: PMC2815515
- DOI: 10.1182/blood-2009-09-243105
Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery
Abstract
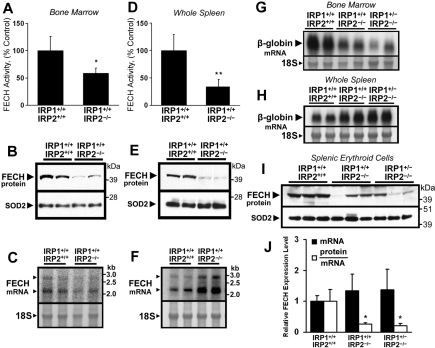
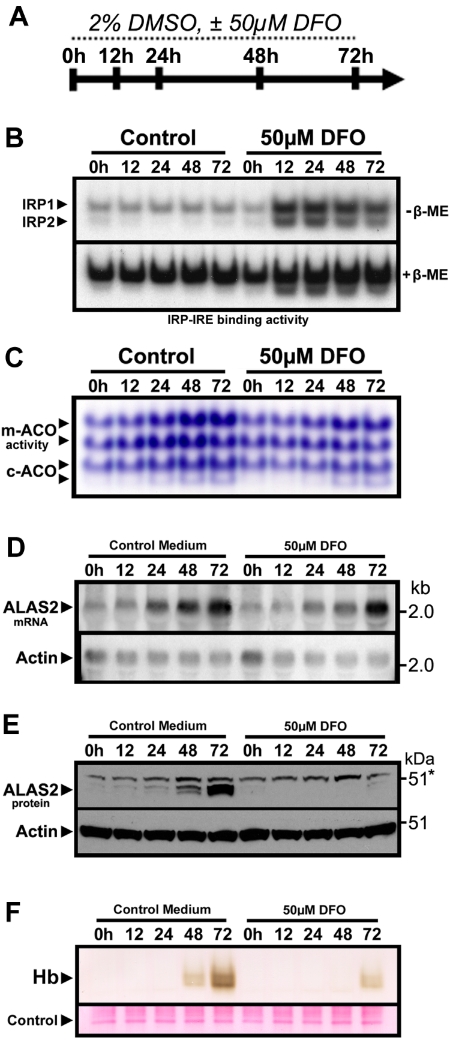
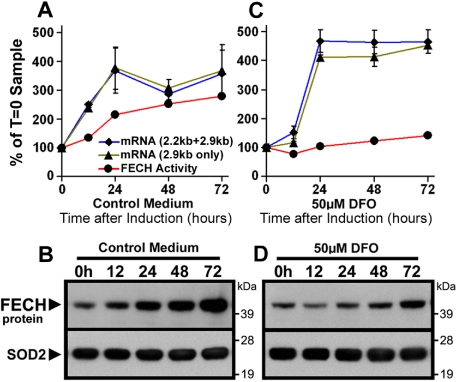
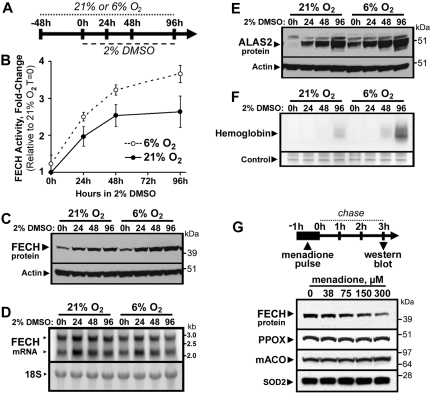
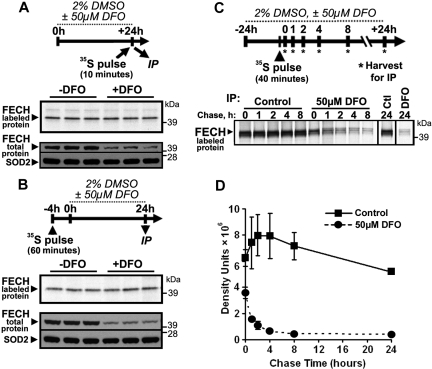
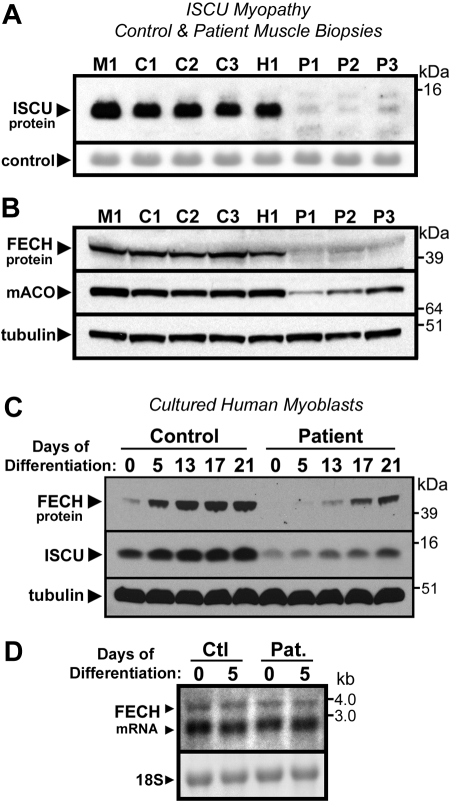
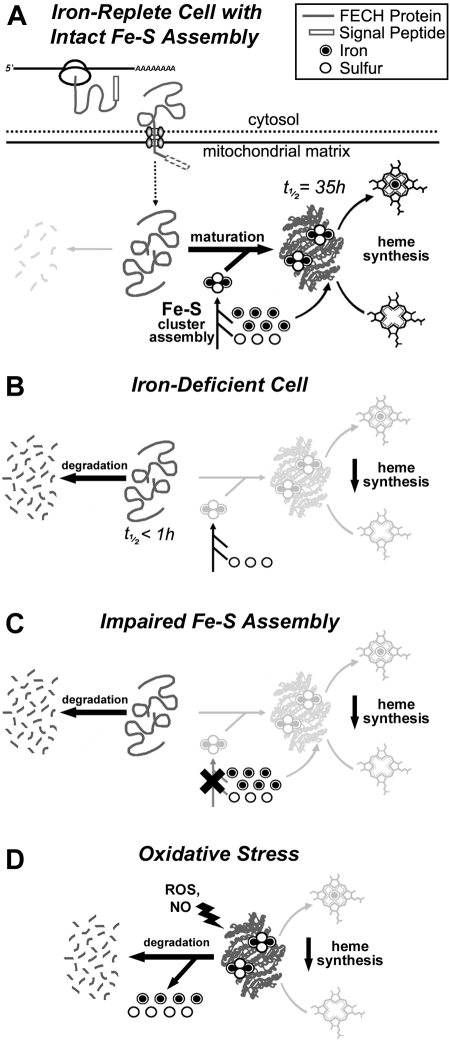
Mammalian ferrochelatase, the terminal enzyme in the heme biosynthetic pathway, possesses an iron-sulfur [2Fe-2S] cluster that does not participate in catalysis. We investigated ferrochelatase expression in iron-deficient erythropoietic tissues of mice lacking iron regulatory protein 2, in iron-deficient murine erythroleukemia cells, and in human patients with ISCU myopathy. Ferrochelatase activity and protein levels were dramatically decreased in Irp2(-/-) spleens, whereas ferrochelatase mRNA levels were increased, demonstrating posttranscriptional regulation of ferrochelatase in vivo. Translation of ferrochelatase mRNA was unchanged in iron-depleted murine erythroleukemia cells, and the stability of mature ferrochelatase protein was also unaffected. However, the stability of newly formed ferrochelatase protein was dramatically decreased during iron deficiency. Ferrochelatase was also severely depleted in muscle biopsies and cultured myoblasts from patients with ISCU myopathy, a disease caused by deficiency of a scaffold protein required for Fe-S cluster assembly. Together, these data suggest that decreased Fe-S cluster availability because of cellular iron depletion or impaired Fe-S cluster assembly causes reduced maturation and stabilization of apo-ferrochelatase, providing a direct link between Fe-S biogenesis and completion of heme biosynthesis. We propose that decreased heme biosynthesis resulting from impaired Fe-S cluster assembly can contribute to the pathogenesis of diseases caused by defective Fe-S cluster biogenesis.
Figures
References
-
- Kikuchi G, Kumar A, Talmage P, Shemin D. The enzymatic synthesis of delta-aminolevulinic acid. J Biol Chem. 1958;233(5):1214–1219. - PubMed
-
- Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006;1763(7):723–736. - PubMed
-
- Nishida G, Labbe RF. Heme biosynthesis; on the incorporation of iron into protoporphyrin. Biochim Biophys Acta. 1959;31(2):519–524. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
