

Enhanced tonic GABAA inhibition in typical absence epilepsy
- PMID: 19966779
- PMCID: PMC2824149
- DOI: 10.1038/nm.2058
Enhanced tonic GABAA inhibition in typical absence epilepsy
Abstract
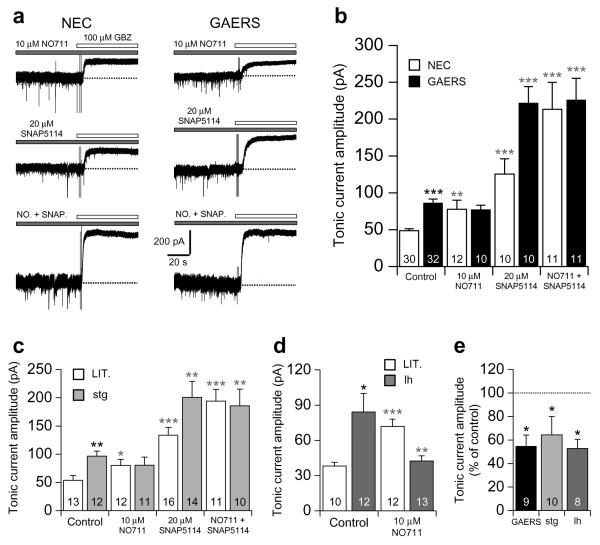
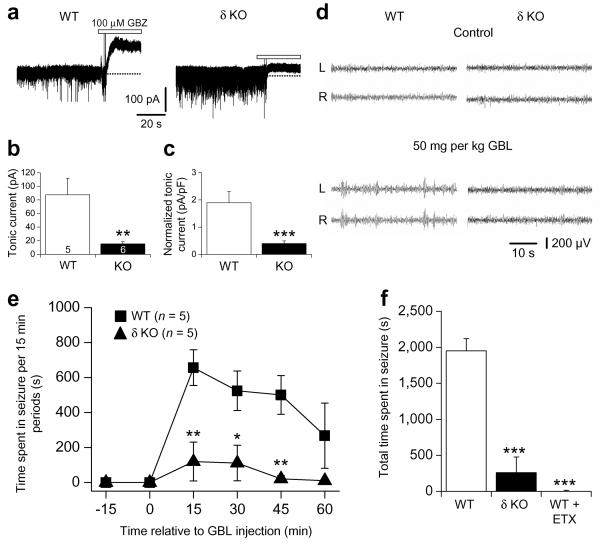
The cellular mechanisms underlying typical absence seizures, which characterize various idiopathic generalized epilepsies, are not fully understood, but impaired gamma-aminobutyric acid (GABA)-ergic inhibition remains an attractive hypothesis. In contrast, we show here that extrasynaptic GABA(A) receptor-dependent 'tonic' inhibition is increased in thalamocortical neurons from diverse genetic and pharmacological models of absence seizures. Increased tonic inhibition is due to compromised GABA uptake by the GABA transporter GAT-1 in the genetic models tested, and GAT-1 is crucial in governing seizure genesis. Extrasynaptic GABA(A) receptors are a requirement for seizures in two of the best characterized models of absence epilepsy, and the selective activation of thalamic extrasynaptic GABA(A) receptors is sufficient to elicit both electrographic and behavioral correlates of seizures in normal rats. These results identify an apparently common cellular pathology in typical absence seizures that may have epileptogenic importance and highlight potential therapeutic targets for the treatment of absence epilepsy.
Figures




References
-
- Commission on Classification and Terminology of the International League Against Epilepsy Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–399. - PubMed
-
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat. Rev. Neurosci. 2002;3:371–382. - PubMed
-
- McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annu. Rev. Physiol. 2001;63:815–846. - PubMed
-
- Blumenfeld H. Cellular and network mechanisms of spike-wave seizures. Epilepsia. 2005;46(Suppl. 9):21–33. - PubMed
-
- von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993;261:361–364. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
