

Evolutionary diversification of an ancient gene family (rhs) through C-terminal displacement
- PMID: 19968874
- PMCID: PMC2935791
- DOI: 10.1186/1471-2164-10-584
Evolutionary diversification of an ancient gene family (rhs) through C-terminal displacement
Abstract
Background: Rhs genes are prominent features of bacterial genomes that have previously been implicated in genomic rearrangements in E. coli. By comparing rhs repertoires across the Enterobacteriaceae, this study provides a robust explanation of rhs diversification and evolution, and a mechanistic model of how rhs diversity is gained and lost.
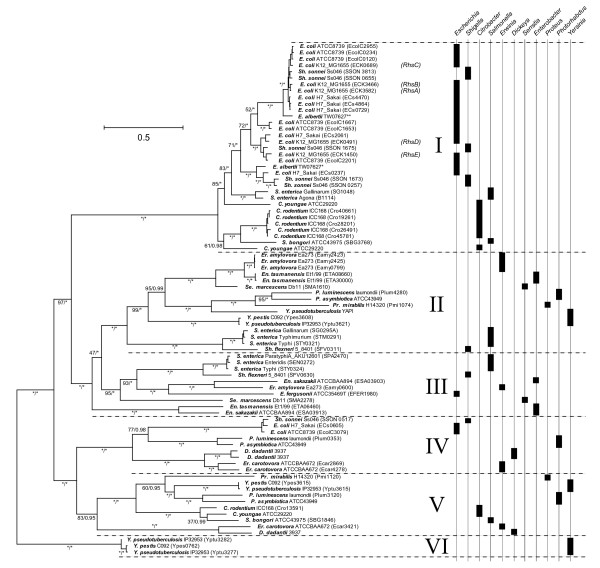
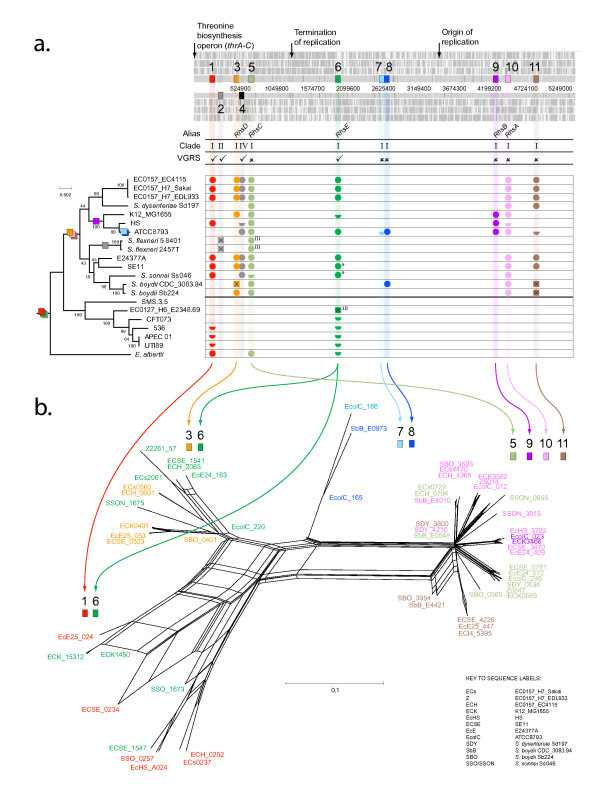
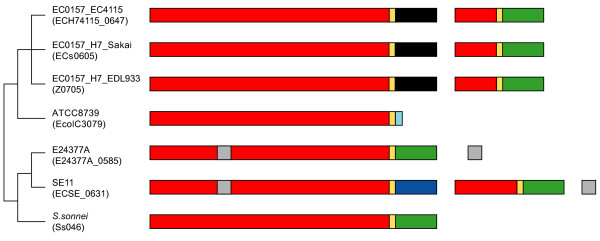
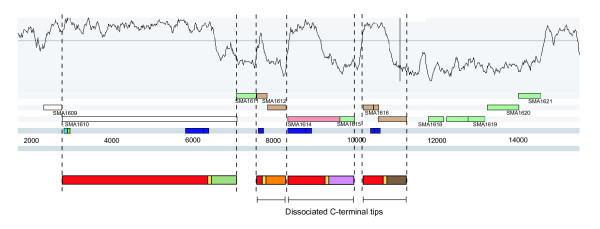
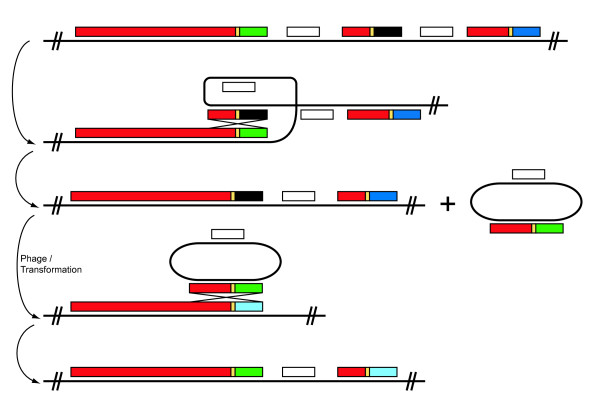
Results: Rhs genes are ubiquitous and comprise six structurally distinct lineages within the Enterobacteriaceae. There is considerable intergenomic variation in rhs repertoire; for instance, in Salmonella enterica, rhs are restricted to mobile elements, while in Escherichia coli one rhs lineage has diversified through transposition as older lineages have been deleted. Overall, comparative genomics reveals frequent, independent gene gains and losses, as well as occasional lateral gene transfer, in different genera. Furthermore, we demonstrate that Rhs 'core' domains and variable C-termini are evolutionarily decoupled, and propose that rhs diversity is driven by homologous recombination with circular intermediates. Existing C-termini are displaced by laterally acquired alternatives, creating long arrays of dissociated 'tips' that characterize the appearance of rhs loci.
Conclusion: Rhs repertoires are highly dynamic among Enterobacterial genomes, due to repeated gene gains and losses. In contrast, the primary structures of Rhs genes are evolutionarily conserved, indicating that rhs sequence diversity is driven, not by rapid mutation, but by the relatively slow evolution of novel core/tip combinations. Hence, we predict that a large pool of dissociated rhs C-terminal tips exists episomally and these are potentially transmitted across taxonomic boundaries.
Figures

Similar articles
-
Rhs elements of Escherichia coli: a family of genetic composites each encoding a large mosaic protein.Mol Microbiol. 1994 Jun;12(6):865-71. doi: 10.1111/j.1365-2958.1994.tb01074.x. Mol Microbiol. 1994. PMID: 7934896 Review.
-
Stratified reconstruction of ancestral Escherichia coli diversification.BMC Genomics. 2019 Dec 5;20(1):936. doi: 10.1186/s12864-019-6346-1. BMC Genomics. 2019. PMID: 31805853 Free PMC article.
-
Phylogenomics and comparative genomic studies delineate six main clades within the family Enterobacteriaceae and support the reclassification of several polyphyletic members of the family.Infect Genet Evol. 2017 Oct;54:108-127. doi: 10.1016/j.meegid.2017.06.024. Epub 2017 Jun 26. Infect Genet Evol. 2017. PMID: 28658607
-
Rhs elements comprise three subfamilies which diverged prior to acquisition by Escherichia coli.J Bacteriol. 1998 Aug;180(16):4102-10. doi: 10.1128/JB.180.16.4102-4110.1998. J Bacteriol. 1998. PMID: 9696756 Free PMC article.
-
Large genomic sequence repetitions in bacteria: lessons from rRNA operons and Rhs elements.Res Microbiol. 1999 Nov-Dec;150(9-10):665-74. doi: 10.1016/s0923-2508(99)00125-4. Res Microbiol. 1999. PMID: 10673005 Review.
Cited by
-
Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics.Biol Direct. 2012 Jun 25;7:18. doi: 10.1186/1745-6150-7-18. Biol Direct. 2012. PMID: 22731697 Free PMC article.
-
Comparative genomics of type VI secretion systems in strains of Pantoea ananatis from different environments.BMC Genomics. 2014 Feb 26;15:163. doi: 10.1186/1471-2164-15-163. BMC Genomics. 2014. PMID: 24571088 Free PMC article.
-
Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems.PLoS Genet. 2011 Aug;7(8):e1002217. doi: 10.1371/journal.pgen.1002217. Epub 2011 Aug 4. PLoS Genet. 2011. PMID: 21829394 Free PMC article.
-
Sequential displacement of Type VI Secretion System effector genes leads to evolution of diverse immunity gene arrays in Vibrio cholerae.Sci Rep. 2017 Mar 22;7:45133. doi: 10.1038/srep45133. Sci Rep. 2017. PMID: 28327641 Free PMC article.
-
Intraspecies Competition in Serratia marcescens Is Mediated by Type VI-Secreted Rhs Effectors and a Conserved Effector-Associated Accessory Protein.J Bacteriol. 2015 Jul;197(14):2350-60. doi: 10.1128/JB.00199-15. Epub 2015 May 4. J Bacteriol. 2015. PMID: 25939831 Free PMC article.
References
-
- Lawrence JG, Retchless AC. The interplay of homologous recombination and horizontal gene transfer in bacterial speciation. Methods Mol Biol. 2009;532:29–53. full_text. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
