

Genes and gene expression modules associated with caloric restriction and aging in the laboratory mouse
- PMID: 19968875
- PMCID: PMC2795771
- DOI: 10.1186/1471-2164-10-585
Genes and gene expression modules associated with caloric restriction and aging in the laboratory mouse
Abstract
Background: Caloric restriction (CR) counters deleterious effects of aging and, for most mouse genotypes, increases mean and maximum lifespan. Previous analyses of microarray data have identified gene expression responses to CR that are shared among multiple mouse tissues, including the activation of anti-oxidant, tumor suppressor and anti-inflammatory pathways. These analyses have provided useful research directions, but have been restricted to a limited number of tissues, and have focused on individual genes, rather than whole-genome transcriptional networks. Furthermore, CR is thought to oppose age-associated gene expression patterns, but detailed statistical investigations of this hypothesis have not been carried out.
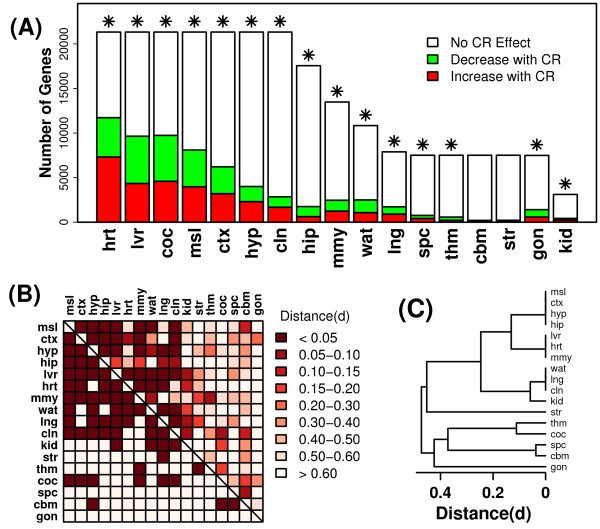
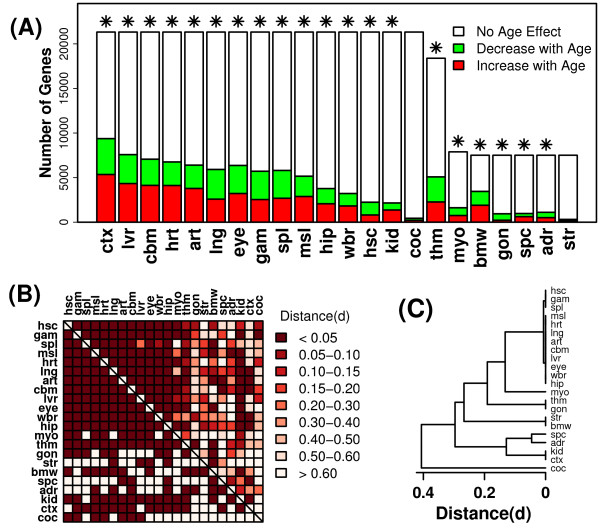
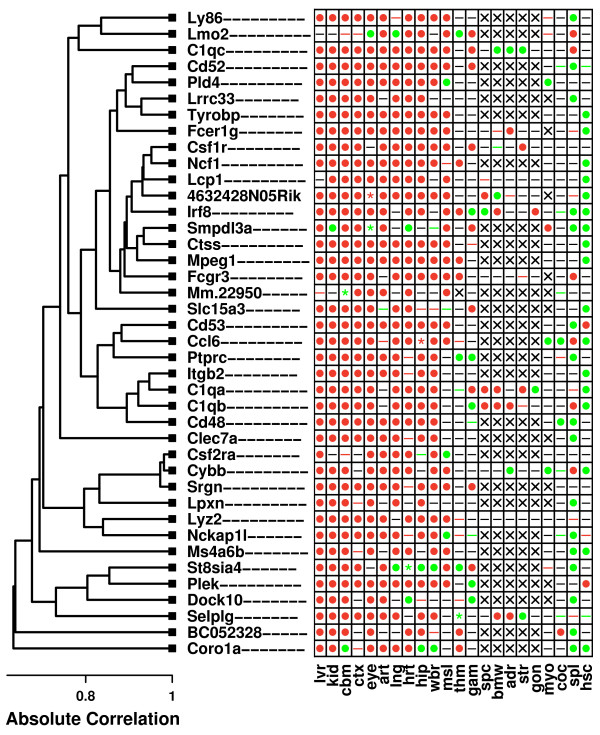
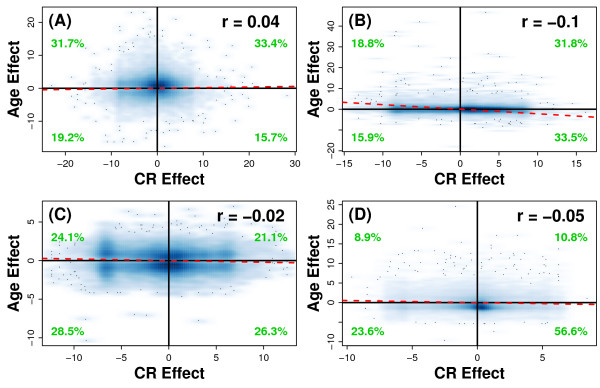
Results: Systemic effects of CR and aging were identified by examining transcriptional responses to CR in 17 mouse tissue types, as well as responses to aging in 22 tissues. CR broadly induced the expression of genes known to inhibit oxidative stress (e.g., Mt1, Mt2), inflammation (e.g., Nfkbia, Timp3) and tumorigenesis (e.g., Txnip, Zbtb16). Additionally, a network-based investigation revealed that CR regulates a large co-expression module containing genes associated with the metabolism and splicing of mRNA (e.g., Cpsf6, Sfpq, Sfrs18). The effects of aging were, to a considerable degree, similar among groups of co-expressed genes. Age-related gene expression patterns characteristic of most mouse tissues were identified, including up regulation of granulin (Grn) and secreted phosphoprotein 1 (Spp1). The transcriptional association between CR and aging varied at different levels of analysis. With respect to gene subsets associated with certain biological processes (e.g., immunity and inflammation), CR opposed age-associated expression patterns. However, among all genes, global transcriptional effects of CR were only weakly related to those of aging.
Conclusion: The study of aging, and of interventions thought to combat aging, has much to gain from data-driven and unbiased genomic investigations. Expression patterns identified in this analysis characterize a generalized response of mammalian cells to CR and/or aging. These patterns may be of importance in determining effects of CR on overall lifespan, or as factors that underlie age-related disease. The association between CR and aging warrants further study, but most evidence indicates that CR does not induce a genome-wide "reversal" of age-associated gene expression patterns.
Figures







References
-
- Weindruch R, Walford RL, Fligiel S, Guthrie D. The Retardation of Aging in Mice by Dietary Restriciton: Longevity, Cancer, Immunity and Lifetime Energy Intake. J Nutrition. 1986;116:641–54. - PubMed
-
- Heilbronn LK, Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr. 2003;78:361–9. - PubMed
-
- Lane MA, Roth GS, Ingram DK. Caloric restriction mimetics: a novel approach for biogerontology. Methods Mol Biol. 2007;371:143–9. full_text. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
