

Transforming growth factor beta1 inhibits cystic fibrosis transmembrane conductance regulator-dependent cAMP-stimulated alveolar epithelial fluid transport via a phosphatidylinositol 3-kinase-dependent mechanism
- PMID: 19996317
- PMCID: PMC2836032
- DOI: 10.1074/jbc.M109.036731
Transforming growth factor beta1 inhibits cystic fibrosis transmembrane conductance regulator-dependent cAMP-stimulated alveolar epithelial fluid transport via a phosphatidylinositol 3-kinase-dependent mechanism
Abstract
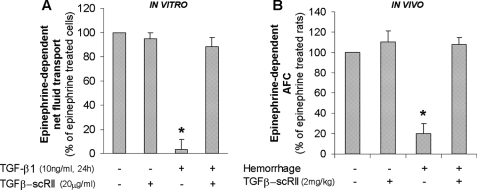
Exogenous or endogenous beta(2)-adrenergic receptor agonists enhance alveolar epithelial fluid transport via a cAMP-dependent mechanism that protects the lungs from alveolar flooding in acute lung injury. However, impaired alveolar fluid clearance is present in most of the patients with acute lung injury and is associated with increased mortality, although the mechanisms responsible for this inhibition of the alveolar epithelial fluid transport are not completely understood. Here, we found that transforming growth factor beta1 (TGF-beta1), a critical mediator of acute lung injury, inhibits beta(2)-adrenergic receptor agonist-stimulated vectorial fluid and Cl(-) transport across primary rat and human alveolar epithelial type II cell monolayers. This inhibition is due to a reduction in the cystic fibrosis transmembrane conductance regulator activity and biosynthesis mediated by a phosphatidylinositol 3-kinase (PI3K)-dependent heterologous desensitization and down-regulation of the beta(2)-adrenergic receptors. Consistent with these in vitro results, inhibition of the PI3K pathway or pretreatment with soluble chimeric TGF-beta type II receptor restored beta(2)-adrenergic receptor agonist-stimulated alveolar epithelial fluid transport in an in vivo model of acute lung injury induced by hemorrhagic shock in rats. The results demonstrate a novel role for TGF-beta1 in impairing the beta- adrenergic agonist-stimulated alveolar fluid clearance in acute lung injury, an effect that could be corrected by using PI3K inhibitors that are safe to use in humans.
Figures






Similar articles
-
IL-8 inhibits cAMP-stimulated alveolar epithelial fluid transport via a GRK2/PI3K-dependent mechanism.FASEB J. 2013 Mar;27(3):1095-106. doi: 10.1096/fj.12-219295. Epub 2012 Dec 6. FASEB J. 2013. PMID: 23221335 Free PMC article.
-
Synergistic Inhibition of β2-adrenergic Receptor-mediated Alveolar Epithelial Fluid Transport by Interleukin-8 and Transforming Growth Factor-β.Anesthesiology. 2015 May;122(5):1084-92. doi: 10.1097/ALN.0000000000000595. Anesthesiology. 2015. PMID: 25591042
-
Transforming growth factor-β1 and cigarette smoke inhibit the ability of β2-agonists to enhance epithelial permeability.Am J Respir Cell Mol Biol. 2015 Jan;52(1):65-74. doi: 10.1165/rcmb.2013-0538OC. Am J Respir Cell Mol Biol. 2015. PMID: 24978189 Free PMC article.
-
Effects of beta2-adrenergic receptor overexpression on alveolar epithelial active transport.J Allergy Clin Immunol. 2002 Dec;110(6 Suppl):S242-6. doi: 10.1067/mai.2002.129706. J Allergy Clin Immunol. 2002. PMID: 12464931 Review.
-
Alveolar epithelium: role in lung fluid balance and acute lung injury.Proc Am Thorac Soc. 2005;2(3):206-13. doi: 10.1513/pats.200501-009AC. Proc Am Thorac Soc. 2005. PMID: 16222039 Review.
Cited by
-
Transforming growth factor-β1 impairs CFTR-mediated anion secretion across cultured porcine vas deferens epithelial monolayer via the p38 MAPK pathway.Am J Physiol Cell Physiol. 2013 Oct 15;305(8):C867-76. doi: 10.1152/ajpcell.00121.2013. Epub 2013 Jul 31. Am J Physiol Cell Physiol. 2013. PMID: 23903699 Free PMC article.
-
Overexpression of TGF-β1 induces renal fibrosis and accelerates the decline in kidney function in polycystic kidney disease.Am J Physiol Renal Physiol. 2020 Dec 1;319(6):F1135-F1148. doi: 10.1152/ajprenal.00366.2020. Epub 2020 Nov 9. Am J Physiol Renal Physiol. 2020. PMID: 33166182 Free PMC article.
-
TGF-β-induced IL-6 prevents development of acute lung injury in influenza A virus-infected F508del CFTR-heterozygous mice.Am J Physiol Lung Cell Mol Physiol. 2015 Jun 1;308(11):L1136-44. doi: 10.1152/ajplung.00078.2015. Epub 2015 Apr 3. Am J Physiol Lung Cell Mol Physiol. 2015. PMID: 25840995 Free PMC article.
-
TGF-β1 Augments the Apical Membrane Abundance of Lemur Tyrosine Kinase 2 to Inhibit CFTR-Mediated Chloride Transport in Human Bronchial Epithelia.Front Cell Dev Biol. 2020 Feb 7;8:58. doi: 10.3389/fcell.2020.00058. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32117984 Free PMC article.
-
Neuronal Wiskott-Aldrich syndrome protein regulates TGF-β1-mediated lung vascular permeability.FASEB J. 2016 Jul;30(7):2557-69. doi: 10.1096/fj.201600102R. Epub 2016 Mar 29. FASEB J. 2016. PMID: 27025963 Free PMC article.
References
-
- Rubenfeld G. D., Caldwell E., Peabody E., Weaver J., Martin D. P., Neff M., Stern E. J., Hudson L. D. (2005) N. Engl. J. Med. 353, 1685–1693 - PubMed
-
- Ware L. B., Matthay M. A. (2000) N. Engl. J. Med. 342, 1334–1349 - PubMed
-
- Ware L. B., Matthay M. A. (2001) Am. J. Respir. Crit. Care Med. 163, 1376–1383 - PubMed
-
- Berthiaume Y. (1991) J. Appl. Physiol. 70, 2490–2497 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
