

Isoflurane postconditioning protects against reperfusion injury by preventing mitochondrial permeability transition by an endothelial nitric oxide synthase-dependent mechanism
- PMID: 19996950
- PMCID: PMC4374483
- DOI: 10.1097/ALN.0b013e3181c4a607
Isoflurane postconditioning protects against reperfusion injury by preventing mitochondrial permeability transition by an endothelial nitric oxide synthase-dependent mechanism
Abstract
Background: The role of endothelial nitric oxide synthase (eNOS) in isoflurane postconditioning (IsoPC)-elicited cardioprotection is poorly understood. The authors addressed this issue using eNOS mice.
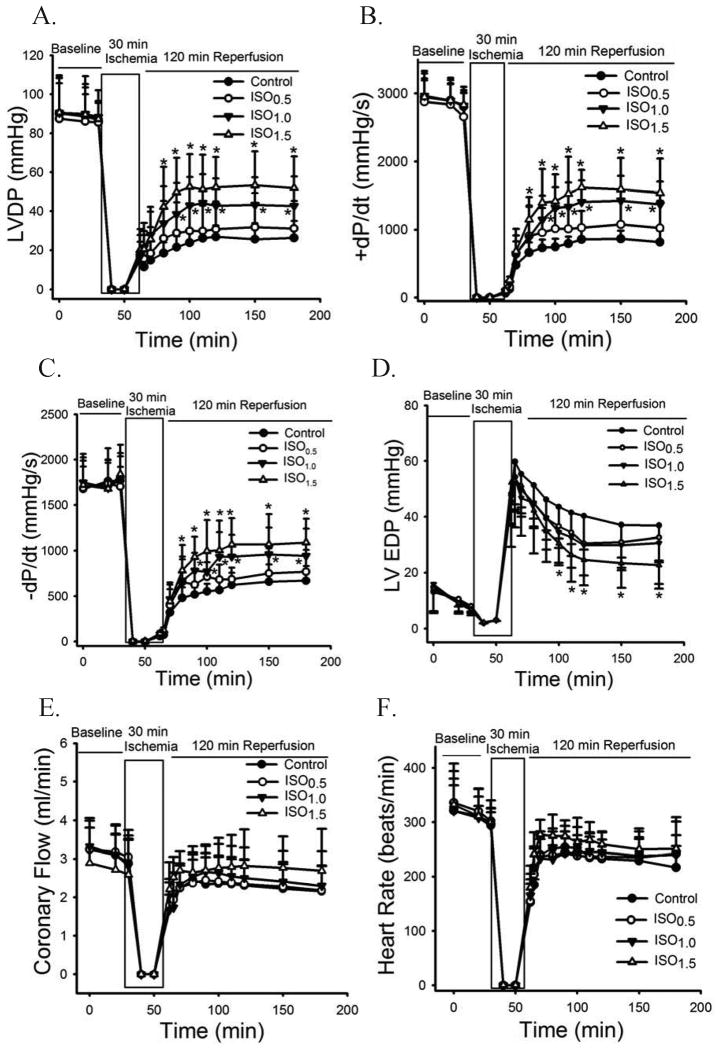
Methods: In vivo or Langendorff-perfused mouse hearts underwent 30 min of ischemia followed by 2 h of reperfusion in the presence and absence of postconditioning produced with isoflurane 5 min before and 3 min after reperfusion. Ca+-induced mitochondrial permeability transition (MPT) pore opening was assessed in isolated mitochondria. Echocardiography was used to evaluate ventricular function.
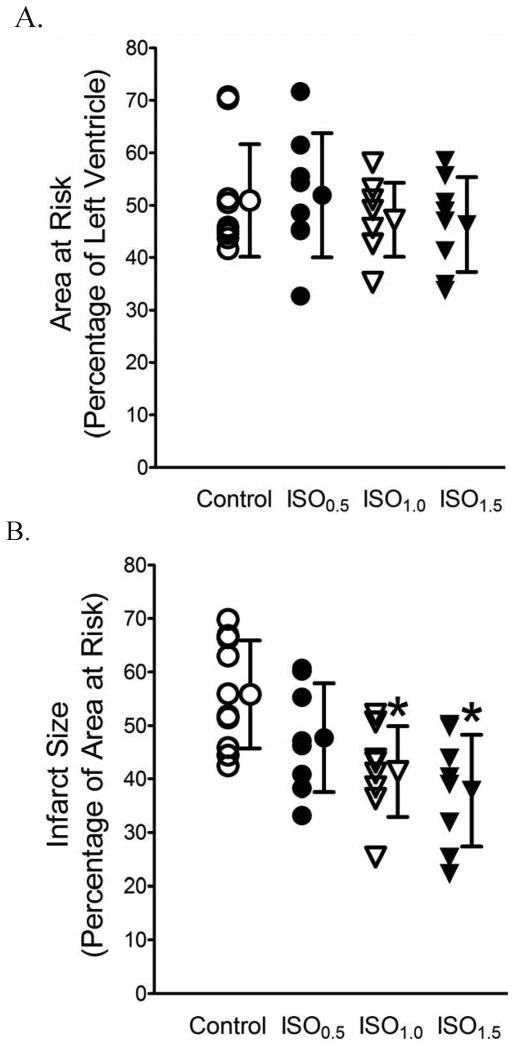
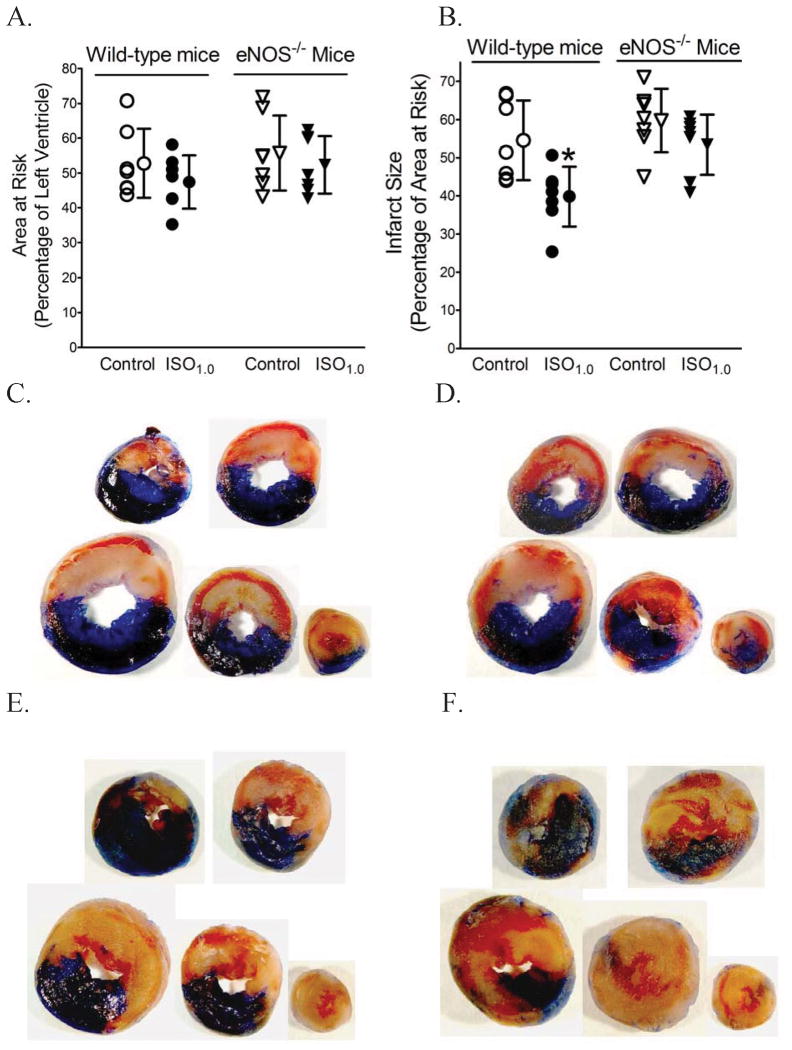
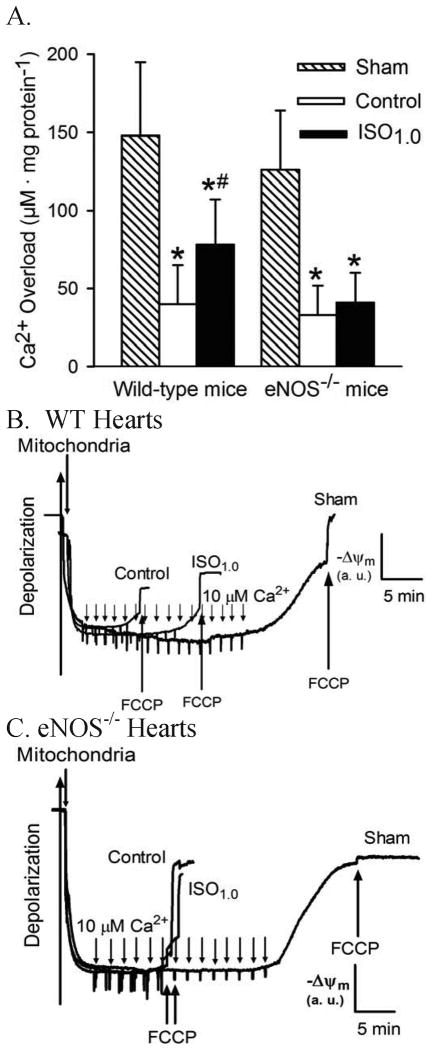
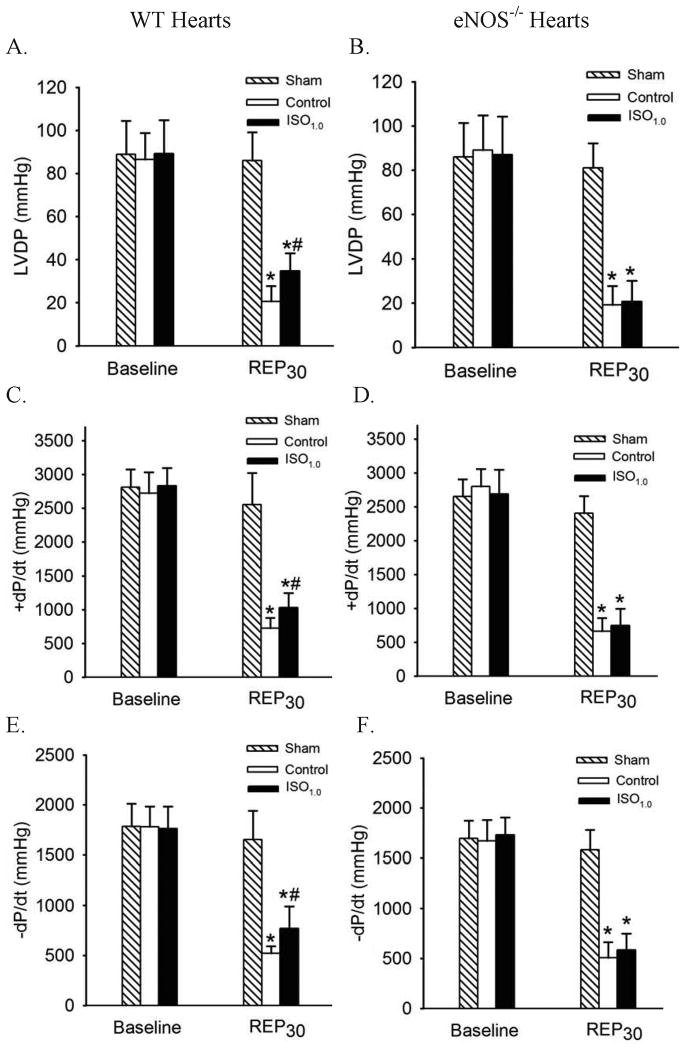
Results: Postconditioning with 0.5, 1.0, and 1.5 minimum alveolar concentrations of isoflurane decreased infarct size from 56 +/- 10% (n = 10) in control to 48 +/- 10%, 41 +/- 8% (n = 8, P < 0.05), and 38 +/- 10% (n = 8, P < 0.05), respectively, and improved cardiac function in wild-type mice. Improvement in cardiac function by IsoPC was blocked by N-nitro-L-arginine methyl ester (a nonselective nitric oxide synthase inhibitor) administered either before ischemia or at the onset of reperfusion. Mitochondria isolated from postconditioned hearts required significantly higher in vitro Ca+ loading than did controls (78 +/- 29 microm vs. 40 +/- 25 microm CaCl2 per milligram of protein, n = 10, P < 0.05) to open the MPT pore. Hearts from eNOS mice displayed no marked differences in infarct size, cardiac function, and sensitivity of MPT pore to Ca+, compared with wild-type hearts. However, IsoPC failed to alter infarct size, cardiac function, or the amount of Ca+ necessary to open the MPT pore in mitochondria isolated from the eNOS hearts compared with control hearts.
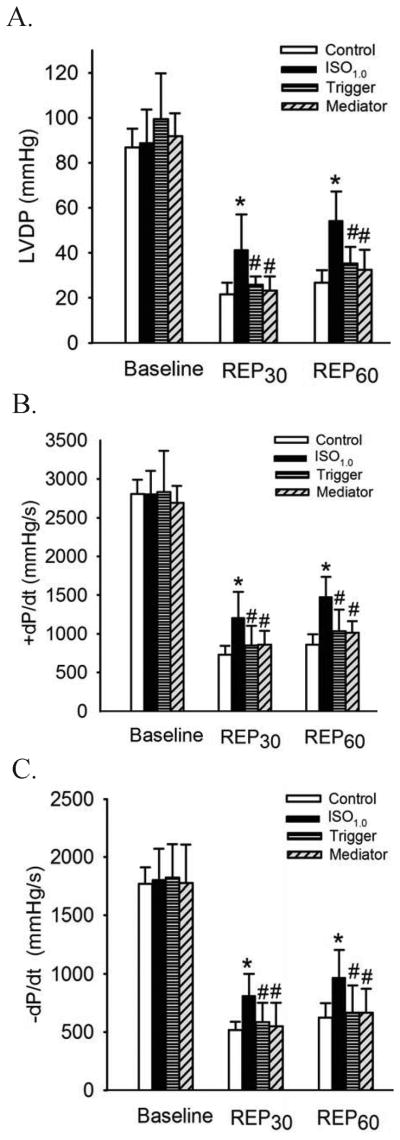
Conclusions: IsoPC protects mouse hearts from reperfusion injury by preventing MPT pore opening in an eNOS-dependent manner. Nitric oxide functions as both a trigger and a mediator of cardioprotection produced by IsoPC.
Figures

Similar articles
-
MicroRNA-21 Mediates Isoflurane-induced Cardioprotection against Ischemia-Reperfusion Injury via Akt/Nitric Oxide Synthase/Mitochondrial Permeability Transition Pore Pathway.Anesthesiology. 2015 Oct;123(4):786-798. doi: 10.1097/ALN.0000000000000807. Anesthesiology. 2015. PMID: 26259139 Free PMC article.
-
Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3beta.Anesthesiology. 2005 Nov;103(5):987-95. doi: 10.1097/00000542-200511000-00013. Anesthesiology. 2005. PMID: 16249673
-
Evidence that hydroxysafflor yellow A protects the heart against ischaemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening.Clin Exp Pharmacol Physiol. 2008 Feb;35(2):211-6. doi: 10.1111/j.1440-1681.2007.04814.x. Epub 2007 Oct 17. Clin Exp Pharmacol Physiol. 2008. PMID: 17941891
-
The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection.J Mol Cell Cardiol. 2015 Jan;78:100-6. doi: 10.1016/j.yjmcc.2014.09.023. Epub 2014 Sep 28. J Mol Cell Cardiol. 2015. PMID: 25268651 Free PMC article. Review.
-
The mitochondrial permeability transition pore and ischemia-reperfusion injury.Basic Res Cardiol. 2009 Mar;104(2):181-8. doi: 10.1007/s00395-009-0004-8. Epub 2009 Feb 26. Basic Res Cardiol. 2009. PMID: 19242640 Free PMC article. Review.
Cited by
-
Cardiomyocyte GTP Cyclohydrolase 1 Protects the Heart Against Diabetic Cardiomyopathy.Sci Rep. 2016 Jun 13;6:27925. doi: 10.1038/srep27925. Sci Rep. 2016. PMID: 27295516 Free PMC article.
-
Role of Endothelial Nitric Oxide Synthase in Isoflurane Conditioning-Induced Neurovascular Protection in Subarachnoid Hemorrhage.J Am Heart Assoc. 2020 Oct 20;9(20):e017477. doi: 10.1161/JAHA.120.017477. Epub 2020 Oct 8. J Am Heart Assoc. 2020. PMID: 33030094 Free PMC article.
-
Pharmacological Conditioning of the Heart: An Update on Experimental Developments and Clinical Implications.Int J Mol Sci. 2021 Mar 3;22(5):2519. doi: 10.3390/ijms22052519. Int J Mol Sci. 2021. PMID: 33802308 Free PMC article. Review.
-
Impairment of endothelial-myocardial interaction increases the susceptibility of cardiomyocytes to ischemia/reperfusion injury.PLoS One. 2013 Jul 22;8(7):e70088. doi: 10.1371/journal.pone.0070088. Print 2013. PLoS One. 2013. PMID: 23894596 Free PMC article.
-
Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection.J Mol Cell Cardiol. 2011 Nov;51(5):803-11. doi: 10.1016/j.yjmcc.2011.06.026. Epub 2011 Jul 21. J Mol Cell Cardiol. 2011. PMID: 21791217 Free PMC article.
References
-
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion. Comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–88. - PubMed
-
- Nishino Y, Webb IG, Davidson SM, Ahmed AI, Clark JE, Jacquet S, Shah AM, Miura T, Yellon DM, Avkiran M, Marber MS. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ Res. 2008;103:307–14. - PubMed
-
- Tissier R, Waintraub X, Couvreur N, Gervais M, Bruneval P, Mandet C, Zini R, Enriquez B, Berdeaux A, Ghaleh B. Pharmacological postconditioning with the phytoestrogen genistein. J Mol Cell Cardiol. 2007;42:79–87. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
