

Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure
- PMID: 19997483
- PMCID: PMC2777313
- DOI: 10.1371/journal.pcbi.1000585
Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure
Abstract
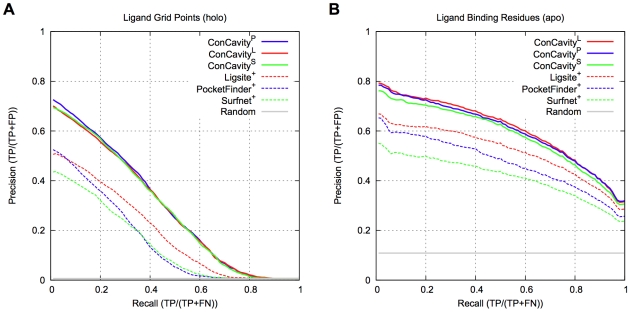
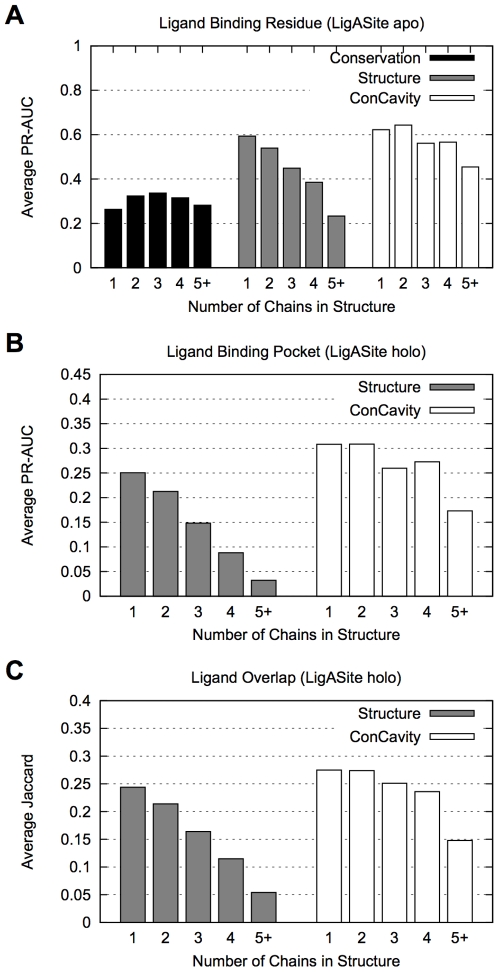
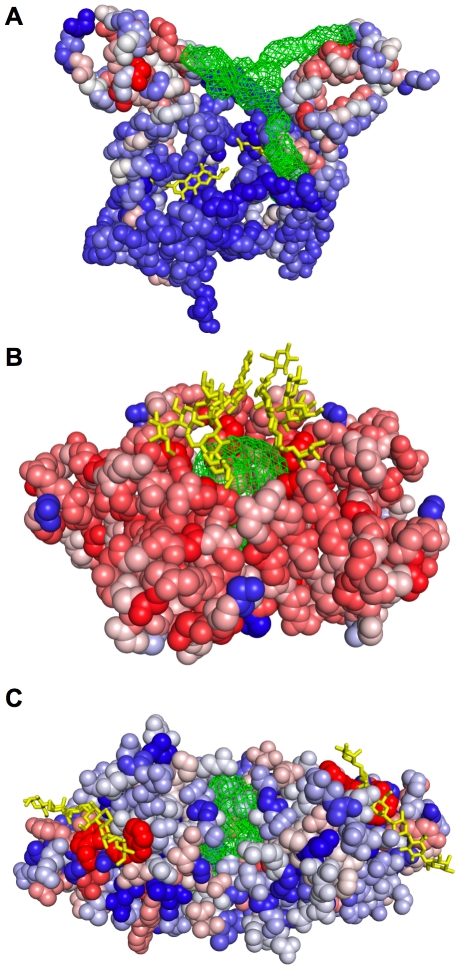
Identifying a protein's functional sites is an important step towards characterizing its molecular function. Numerous structure- and sequence-based methods have been developed for this problem. Here we introduce ConCavity, a small molecule binding site prediction algorithm that integrates evolutionary sequence conservation estimates with structure-based methods for identifying protein surface cavities. In large-scale testing on a diverse set of single- and multi-chain protein structures, we show that ConCavity substantially outperforms existing methods for identifying both 3D ligand binding pockets and individual ligand binding residues. As part of our testing, we perform one of the first direct comparisons of conservation-based and structure-based methods. We find that the two approaches provide largely complementary information, which can be combined to improve upon either approach alone. We also demonstrate that ConCavity has state-of-the-art performance in predicting catalytic sites and drug binding pockets. Overall, the algorithms and analysis presented here significantly improve our ability to identify ligand binding sites and further advance our understanding of the relationship between evolutionary sequence conservation and structural and functional attributes of proteins. Data, source code, and prediction visualizations are available on the ConCavity web site (http://compbio.cs.princeton.edu/concavity/).
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
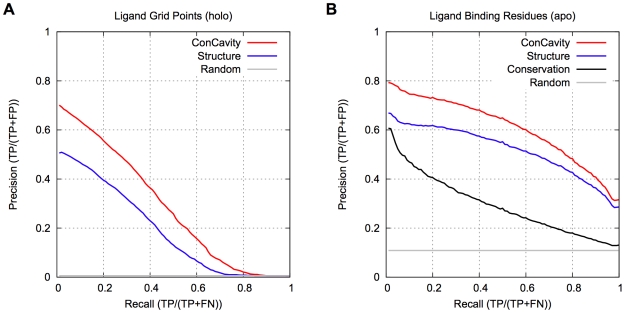
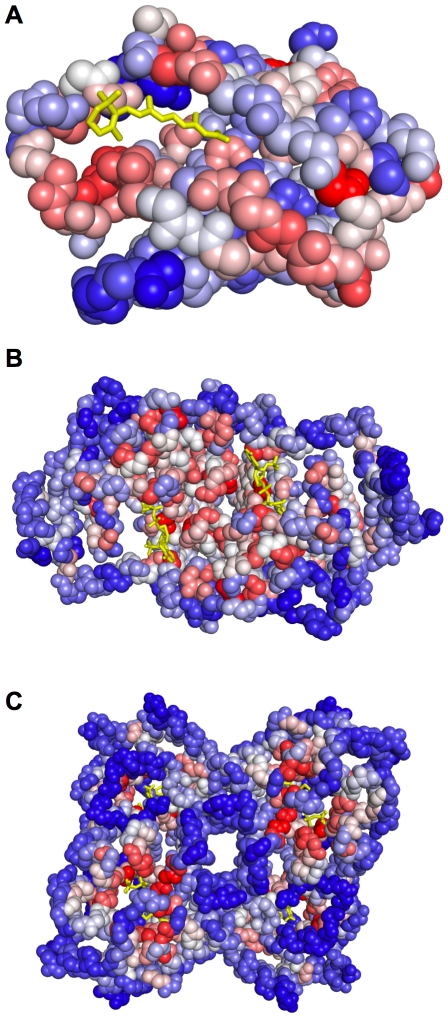
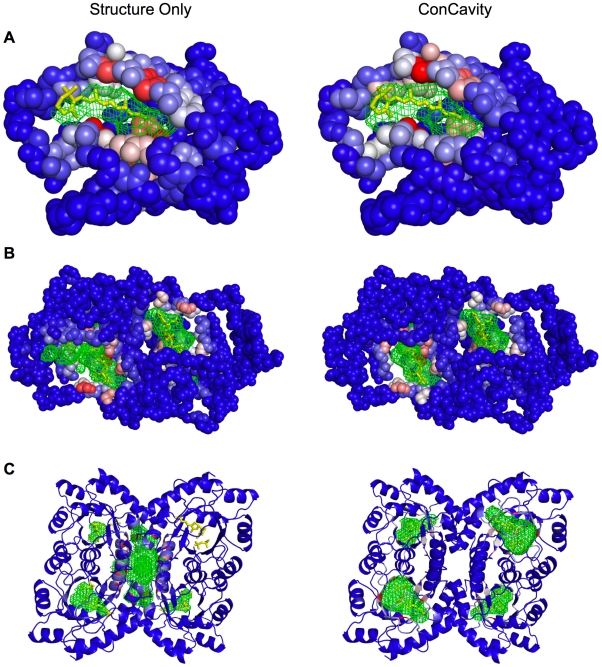
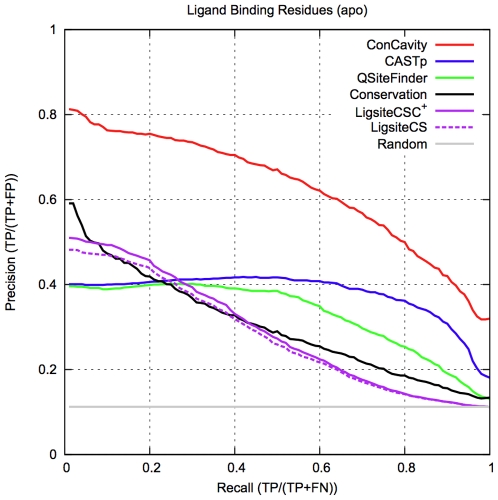
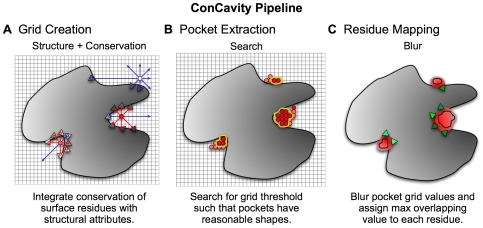
 ) of the pockets, and assign each residue the highest overlapping grid value. Residues near regions of space with very high grid values receive the highest scores.
) of the pockets, and assign each residue the highest overlapping grid value. Residues near regions of space with very high grid values receive the highest scores.Similar articles
-
Predicting functionally important residues from sequence conservation.Bioinformatics. 2007 Aug 1;23(15):1875-82. doi: 10.1093/bioinformatics/btm270. Epub 2007 May 22. Bioinformatics. 2007. PMID: 17519246
-
Predicting protein-ligand binding site using support vector machine with protein properties.IEEE/ACM Trans Comput Biol Bioinform. 2013 Nov-Dec;10(6):1517-29. doi: 10.1109/TCBB.2013.126. IEEE/ACM Trans Comput Biol Bioinform. 2013. PMID: 24407309
-
Improvement in protein functional site prediction by distinguishing structural and functional constraints on protein family evolution using computational design.Nucleic Acids Res. 2005 Oct 13;33(18):5861-7. doi: 10.1093/nar/gki894. Print 2005. Nucleic Acids Res. 2005. PMID: 16224101 Free PMC article.
-
Capturing the geometry, function, and evolution of enzymes with 3D templates.Protein Sci. 2022 Jul;31(7):e4363. doi: 10.1002/pro.4363. Protein Sci. 2022. PMID: 35762726 Free PMC article. Review.
-
FINDSITE: a combined evolution/structure-based approach to protein function prediction.Brief Bioinform. 2009 Jul;10(4):378-91. doi: 10.1093/bib/bbp017. Epub 2009 Mar 26. Brief Bioinform. 2009. PMID: 19324930 Free PMC article. Review.
Cited by
-
Protein-ligand binding region prediction (PLB-SAVE) based on geometric features and CUDA acceleration.BMC Bioinformatics. 2013;14 Suppl 4(Suppl 4):S4. doi: 10.1186/1471-2105-14-S4-S4. Epub 2013 Mar 8. BMC Bioinformatics. 2013. PMID: 23514235 Free PMC article.
-
Exploring functionally related enzymes using radially distributed properties of active sites around the reacting points of bound ligands.BMC Struct Biol. 2012 Apr 26;12:5. doi: 10.1186/1472-6807-12-5. BMC Struct Biol. 2012. PMID: 22536854 Free PMC article.
-
Bioinformatics and variability in drug response: a protein structural perspective.J R Soc Interface. 2012 Jul 7;9(72):1409-37. doi: 10.1098/rsif.2011.0843. Epub 2012 May 2. J R Soc Interface. 2012. PMID: 22552919 Free PMC article. Review.
-
Exploring the landscape of protein-ligand interaction energy using probabilistic approach.J Comput Biol. 2011 Jun;18(6):843-50. doi: 10.1089/cmb.2010.0017. Epub 2010 Nov 20. J Comput Biol. 2011. PMID: 21091064 Free PMC article.
-
Probabilistic Pocket Druggability Prediction via One-Class Learning.Front Pharmacol. 2022 Jun 29;13:870479. doi: 10.3389/fphar.2022.870479. eCollection 2022. Front Pharmacol. 2022. PMID: 35847005 Free PMC article.
References
-
- Capra J, Singh M. Predicting functionally important residues from sequence conservation. Bioinf. 2007;23:1875–1882. - PubMed
-
- Kuznetsov I, Gou Z, Li R, Hwang S. Using evolutionary and structural information to predict DNA-binding sites on DNA-binding proteins. Proteins: Stuct, Func, and Bioinf. 2006;64:19–27. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
